Thu, Jan 29, 2026
Volume 10, Issue 4 (Autumn 2024)
Caspian J Neurol Sci 2024, 10(4): 278-293 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Razmkhah Z, Mosleh H, Aghazadeh-Habashi K, Pouraminaee F, Samadian M, Dadras H, et al . The Role of CSF Biomarkers in Discriminating Between Cerebral Amyloid Angiopathy and Alzheimer’s Disease Patients: Systematic Review of Clinical Studies. Caspian J Neurol Sci 2024; 10 (4) :278-293
URL: http://cjns.gums.ac.ir/article-1-713-en.html
URL: http://cjns.gums.ac.ir/article-1-713-en.html
Zahra Razmkhah1 

 , Haideh Mosleh2 , Komeil Aghazadeh-Habashi3 , Fatemeh Pouraminaee4 , Mohammad Samadian5 , Hadieh Dadras6 , Mahmood Gorjizad7 , Mina Dehghani8 , Kimia Eyvani *9 , Milad Alipour10 , Mahsa Asadi Anar11
, Haideh Mosleh2 , Komeil Aghazadeh-Habashi3 , Fatemeh Pouraminaee4 , Mohammad Samadian5 , Hadieh Dadras6 , Mahmood Gorjizad7 , Mina Dehghani8 , Kimia Eyvani *9 , Milad Alipour10 , Mahsa Asadi Anar11
, Haideh Mosleh2 , Komeil Aghazadeh-Habashi3 , Fatemeh Pouraminaee4 , Mohammad Samadian5 , Hadieh Dadras6 , Mahmood Gorjizad7 , Mina Dehghani8 , Kimia Eyvani *9 , Milad Alipour10 , Mahsa Asadi Anar11
1- Student Research Committee, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran.
2- Hearing Disorder Research Center, Loghman Hakim Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3- Student Research Committee, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.
4- Department of Pathology, Schools of Medicine, Shiraz University of Medical Science, Shiraz, Iran.
5- Skull Base Research Center, Loghman Hakim Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
6- Student Research Committee, School of Medicine, Babol University of Medical Sciences, Babol, Iran.
7- Student Research Committee, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
8- Student Research Committee, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
9- Department of Medicine, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran. ,eyvanikimia@gmail.com
10- Department of Medicine, Faculty of Medicine, Tehran Medical Sciences Branch, Islamic Azad University, Tehran, Iran.
11- Department of Medicine, College of Medicine, University of Arizona, Tucson, United States.
2- Hearing Disorder Research Center, Loghman Hakim Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3- Student Research Committee, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.
4- Department of Pathology, Schools of Medicine, Shiraz University of Medical Science, Shiraz, Iran.
5- Skull Base Research Center, Loghman Hakim Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
6- Student Research Committee, School of Medicine, Babol University of Medical Sciences, Babol, Iran.
7- Student Research Committee, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
8- Student Research Committee, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran.
9- Department of Medicine, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran. ,
10- Department of Medicine, Faculty of Medicine, Tehran Medical Sciences Branch, Islamic Azad University, Tehran, Iran.
11- Department of Medicine, College of Medicine, University of Arizona, Tucson, United States.
Keywords: Cerebral amyloid angiopathy (CAA), Alzheimer disease (AD), Amyloid β (Aβ), Cerebrospinal fluid (CSF)
Full-Text [PDF 2685 kb]
(536 Downloads)
| Abstract (HTML) (1238 Views)
Full-Text: (918 Views)
Introduction
Alzheimer disease (AD), the most predominant cause of dementia, is characterized by advanced memory impairment and cognitive disabilities. Extracellular amyloid β (Aβ) plaques and hyper-phosphorylated tau in the form of intracellular neurofibrillary tangles (NFTs) are the two main pathological hallmarks of AD [1]. Extracellular Aβ peptides are produced directly by the sequential cleavage of the amyloid precursor protein by β-secretase one and γ-secretase, or they can be transferred into the extracellular space from the endosomes, which contain β- and γ-secretase. Post-translational alterations, including phosphorylation, acetylation, ubiquitination, and truncation, generate pathological tau. These alterations often lead to AD-associated tauopathy, characterized by conformational changes in tau protein, its aggregation, and the formation of toxic oligomers and NFTs [2].
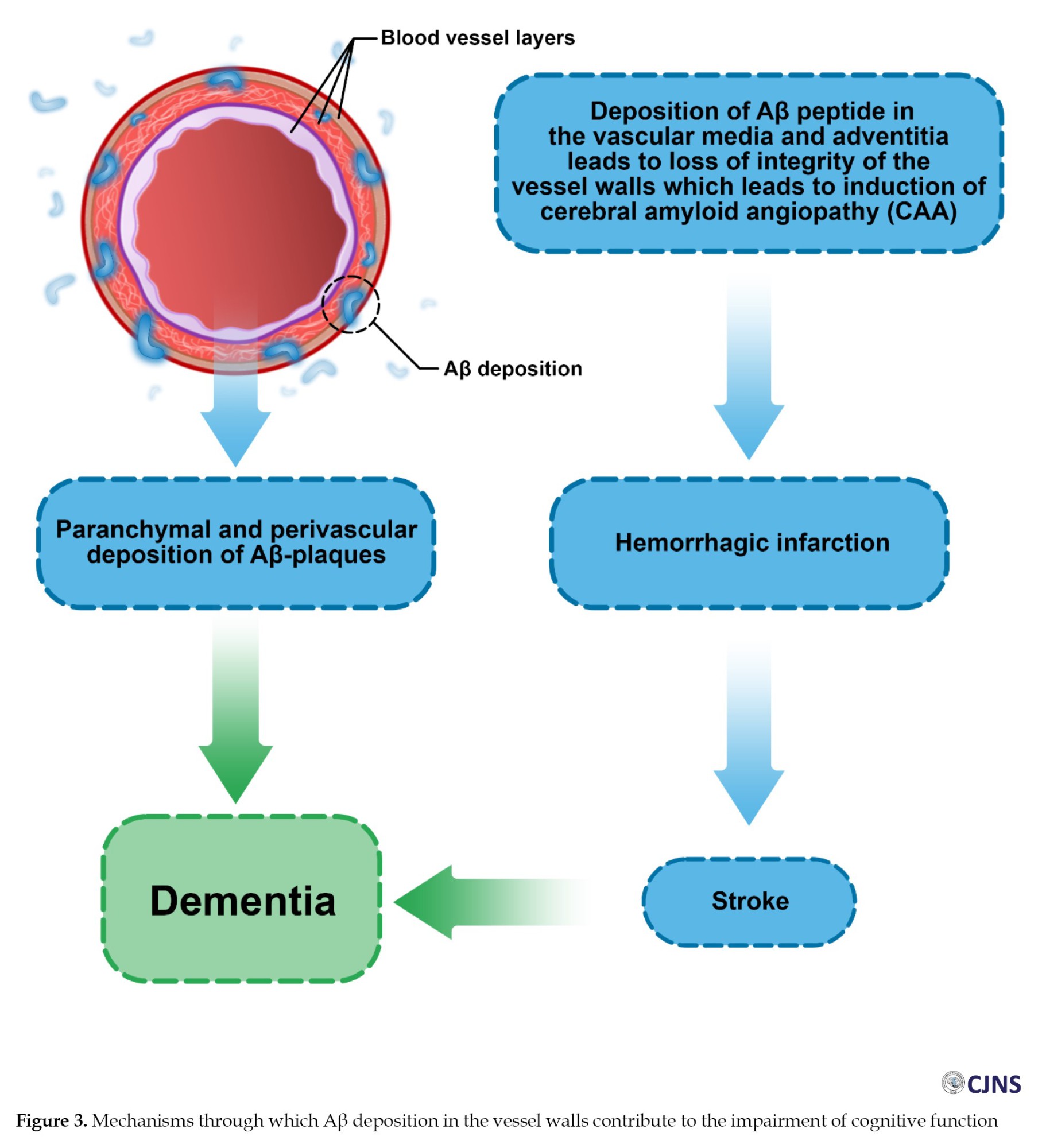
Cerebral amyloid angiopathy (CAA) is a highly prevalent neurological disorder among older people, defined by the accumulation of Aβ peptides within the leptomeninges and walls of small- and medium-size cerebrocortical blood vessels. The deposition of amyloid weakens vasculature and cerebral blood vessel integrity loss, which may manifest in lobar intracerebral hemorrhages (ICH) and cognitive impairment [3]. Aβ peptides participate in the pathogenesis of CAA and are the major components of amyloid plaques found in both cerebral vasculature and brain parenchyma. In vascular deposits, Aβ appears mainly as a 39–40 amino acid peptide (Aβv) in contrast to the 42–43 amino acid peptides that predominate in plaques (Aβp) [4-6]. In CAA, depositions include Aβ40 and Aβ42, with Aβ40 being the dominant form. Increased Aβ40 accumulation is related to the progression of CAA [6-8].
Body fluid biomarkers are typically measured in either cerebrospinal fluid (CSF) or blood due to the ease of collecting these clinical materials. This measure facilitates repeat testing, allowing us to monitor disease-related processes and provide perspectives on disease dynamics [9]. AD biomarkers include CSF levels of total tau (t-tau), phosphorylated tau (p-tau), and Aβ42, which are associated with brain amyloid content and neurodegenerative processes [10, 11]. Changes in Aβ42 concentrations in the CSF (but not the plasma) have been proposed as a possible AD diagnostic criterion [12, 13]. However, increasing evidence indicates that the Aβ42/Aβ40 ratio could benefit the differential diagnosis between AD and other dementias [14]. CSF protein profile in CAA differs from that in AD [10]. The direct evaluation of tau, Aβ peptides, and also other protein markers in the CSF of CAA patients reveals that measurements of t-tau, p-tau181, Aβ40, and Aβ42 could help distinguish CAA and AD patients from controls [14].
Although CAA and AD have numerous molecular similarities, they are not identical [4]. Both are associated with aging, and it has proven difficult to distinguish between vascular brain pathology in CAA and classical AD [15]. CAA can be diagnosed as a standalone disorder or can accompany AD [7]. According to neuropathological studies, more than 90% of AD patients with confirmed plaques and tangles also display signs of CAA, which is caused by Aβ infiltrating into the brain vasculature. However, CAA is also a common condition in non-demented older people, with prevalence rates estimated to range between 20% and 40% [15]. Thus, additional diagnostic criteria are needed to verify the clinical diagnosis due to the pathological and clinical similarities between CAA and AD [16]. Cumulative evidence suggests that CSF biomarkers, t-tau, p-tau, Aβ40, and Aβ42/Aβ40 ratio, can differentiate between CAA and AD [7, 14]. This systematic review aimed to identify all available studies that measured t-tau, p-tau, and or Aβ peptides in the CSF of CAA and AD patients and control subjects. This systematic review aims to establish whether changes in the CSF levels of these proteins could be used as biomarkers, allowing the differential diagnosis between CAA, AD, and control groups. To the best of our knowledge, a systematic review comparing protein biomarkers in the CSF of CAA and AD patients has not been conducted before. Previous systematic reviews have focused on differential diagnosis between AD patients and controls.
Materials and Methods
Literature search strategy
The relevant English articles published from 2009 until 2022 were explored in Medline, Web of Science (WoS), Embase, and Google Scholar. Two authors independently searched using the following key terms and Booleans: ((Alzheimer’s disease) OR (Alzheimer type dementia*) OR (ATD) OR (AD) OR (primary senile degenerative dementia) OR (acute confusional senile dementia) OR (presenile dementia)) AND ((cerebral amyloid angiopathy) OR (CAA) OR (HCHWA) OR (cerebrovascular amyloidosis)) AND ((Aβ40) OR (Aβ42) OR (amyloid-β40) OR (amyloid-β42) OR (tau protein markers) OR (p-tau) OR (t-tau)). The reference lists of all identified studies were also reviewed.
Eligibility criteria
Papers written in English and published in peer-reviewed journals that met all the following inclusion criteria were considered: (1) Observational studies, which (2) report the levels of t-tau, p-tau, Aβ40, Aβ42, or Aβ42/Aβ40 ratio (3) in the CSF of (4) CAA and AD patients, and (5) compare the concentrations of the above proteins in the CSF of CAA and AD patients, or between CAA and or AD patients and a control group. The exclusion criteria were as follows: (1) Non-English studies, (2) animal studies, (3) review articles, conference papers, book chapters, and case reports, (4) studies without randomized sampling, (5) studies reporting the above protein levels only for the AD or CAA patients, (6) studies reporting concentration of the above proteins only in the plasma or the serum of patients, and (7) studies with low-quality scores.
Screening and data extraction
The studies our search assessed were initially screened by their “titles” and “abstracts”. Then, the full texts of selected articles were examined, and eligible articles were included in our study. Relevant data, including authors’ names, year of publication, country of origin, the sample size for the patient and control groups, statistical methods and criteria used, and tau and Aβ concentrations in the CSF, were independently extracted from the included studies by two authors. The results were compared, and any discrepancies were corrected with the help of an additional author.
Results
Study selection
As shown in Figure 1, we extracted 2196 studies for the initial screening after removing duplicated manuscripts.
 From this deduplicated list, 10 studies were chosen for the analysis. The included studies were published between 2009 and 2024 (Table 1).
From this deduplicated list, 10 studies were chosen for the analysis. The included studies were published between 2009 and 2024 (Table 1).
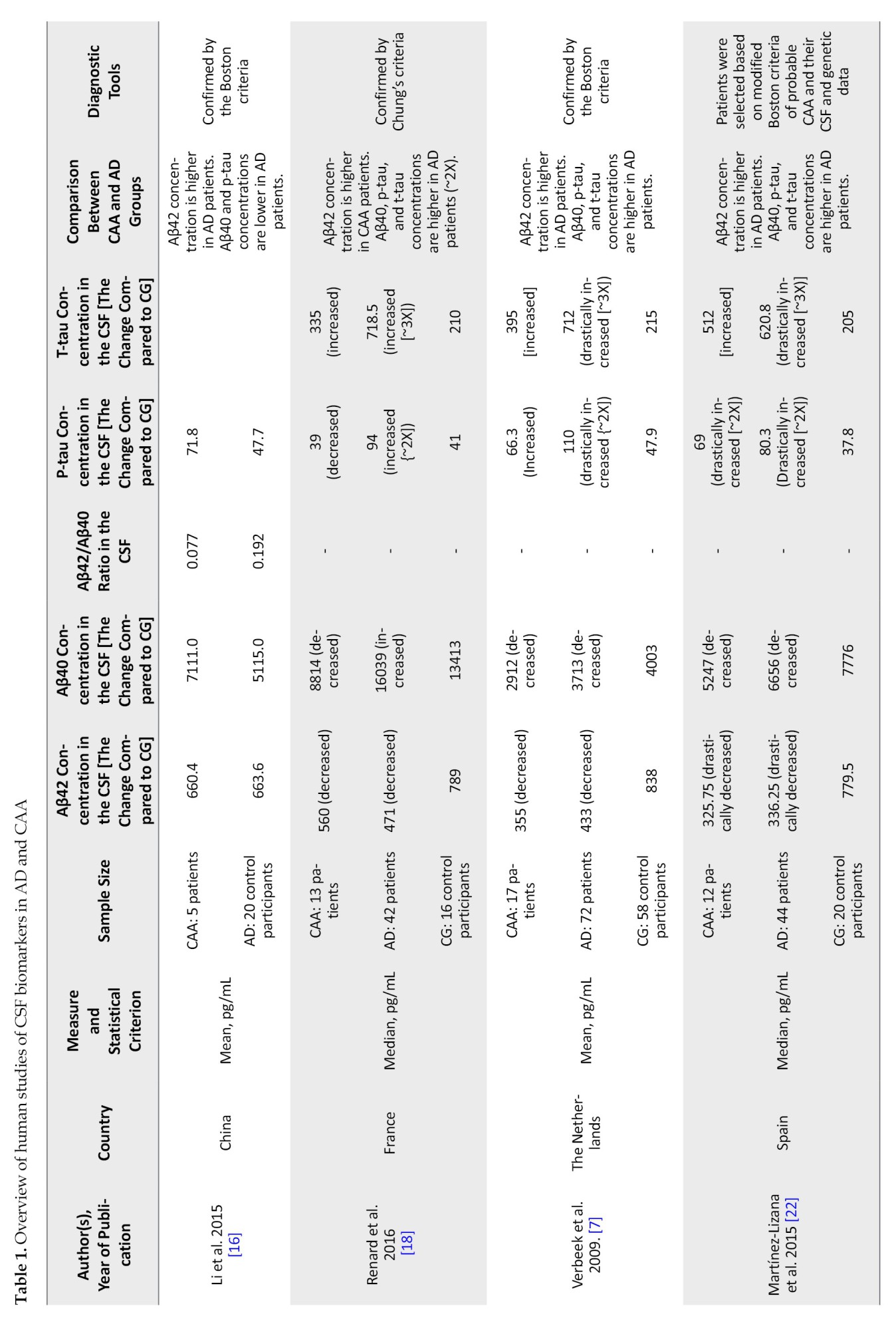

Diagnostic tools
Most studies selected CAA patients based on the imaging results determined by Boston’s probable CAA criteria. One study [17] used neuropathological confirmation for the CAA diagnosis, and two others [10, 18] employed Chung diagnostic criteria.
While most of the included studies identified AD patients based on the diagnostic criteria of probable AD defined by the National Institute of Neurological and Communicative Disorders and Stroke/AD and Related Disorders Association, Renard et al. [18], Margraf et al. [14], Marazuela et al. [17], and Grangeon et al [19] validated AD diagnosis based on the diagnostic criteria of Alzheimer dementia according to the National Institute on Aging and Alzheimer’s Association.
AD patients had a final diagnosis confirmed in all studies based on imaging data, CSF biomarker measurement, and clinical evaluation. Since CAA leads to lobar hemorrhage, vascular dysfunction, and cognitive decline independent of AD patients with CAA were probably clinically distinctive from AD. However, to prevent overlap and exclude patients with mixed CAA and AD pathology, MRI imaging was performed and reviewed by neuroradiologists for the presence of any cerebral microbleeds, cortical, sub-cortical, and lobar hemorrhagic lesions, and evidence of previous infarction. Specimens from individuals exhibiting these characteristics were excluded from the study. Tau and Aβ levels in the CSF samples were used to identify the probable CAA, AD groups, and healthy individuals. Subjects with evidence or a history of any central nervous system disease, neurodegenerative disease, stroke, arteriovenous malformation, recent brain trauma, or tumor were excluded from the control group.
Contribution of Aβ species to CAA and AD
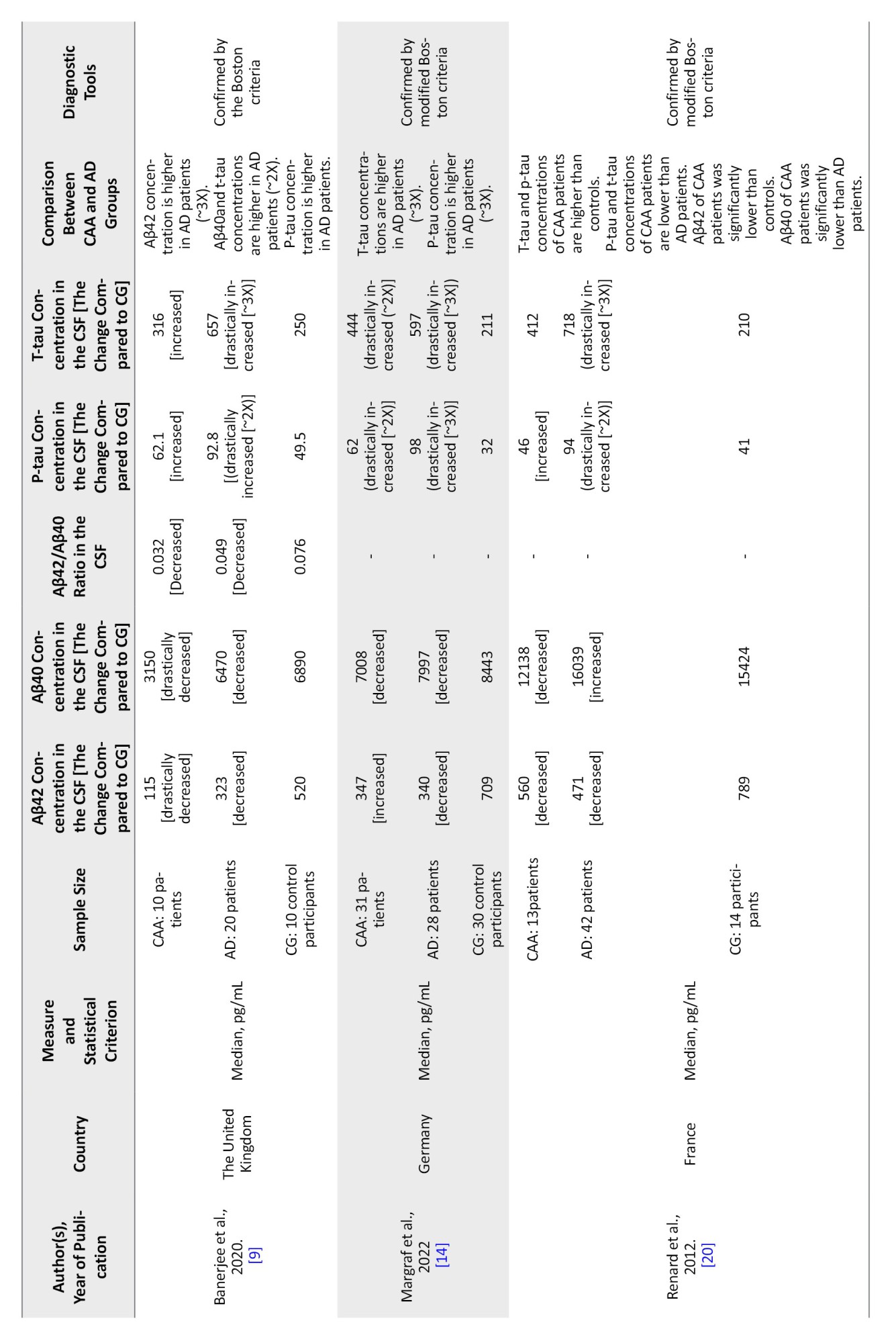
Ten articles analyzing CSF Aβ40 and Aβ42 levels were included in this review. In the study conducted by Li et al. [16] in China, CSF levels of Aβ40 and Aβ42 in 5 CAA patients were compared to those in 20 AD patients. This study had no control group that could be compared with the CAA and or AD groups. All 5 CAA patients, as well as 12 AD patients, were males. The mean age in the CAA and AD groups was 74.4 and 67.3 years, respectively. The Mean±SD CSF levels of Aβ40 in the CAA group were higher than that in the AD group (CAA: 7111 pg/mL; AD: 5115±2931 pg/mL). However, this difference did not seem statistically significant (P=0.15). Statistical t-test was used to compare the means of Aβ40 levels between the two groups. There was no difference between the concentration of Aβ42 in the CSF from CAA and AD patients (CAA: 660.4 pg/mL; AD: 663.6 pg/mL; P=0.99). In 2016, Renard et al. [10] published a prospective study on 9 patients with CAA-related inflammation in the acute phase. They compared the Aβ protein levels in CSF of these patients to those of 10 CAA patients not displaying acute inflammation, 42 AD patients, 3 patients with primary angiitis of the central nervous system, and 14 controls. Based on the data provided in this study, the CSF concentration of Aβ40 in AD patients was higher than in CAA group and the control subjects. Also, the CAA group was not significantly different from the controls. Aβ42 was higher in CAA (without inflammation) compared to AD patients, though both groups showed a decrease in this biomarker compared to control subjects. In another study, Renard et al. [20] measured CSF biomarkers in 13 CAA patients, 42 AD patients, and 14 controls. They observed that CSF Aβ40 levels in CAA patients decreased significantly compared with the AD patients (CAA: 12138 pg/mL; AD: 16039 pg/mL; P=0.0014). However, the difference between the CSF Aβ40 levels in CAA and control groups was not significant (control: 15424 pg/mL; P=0.20 compared to the CAA group, H-score=1.59). They also observed that CSF Aβ42 levels in the CAA group were significantly lower than in controls (CAA: 560 pg/mL; control: 789 pg/mL; P=0.027). However, the difference between the CAA and the AD group was not significant (AD: 471 pg/mL; P=0.494 compared to the CAA group). Later, in another study, Renard et al. [10] recorded significantly lowered CSF Aβ40 levels (P<0.001) but not significantly lowered Aβ42 levels (P=0.35) in the CAA patient group compared with the AD group. In a prior systematic review and meta-analysis by Charidimou et al. [21], three studies conducted by Renard et al. [18], Verbeek et al. [7], and Martínez-Lizana et al. [22] were investigated. These studies were also included in our work and explained in detail below.
Renard et al. [18] studied 13 CAA patients with lobar hematoma and 42 AD patients compared to 16 control subjects. The CSF levels of Aβ40 and Aβ42 were lower in CAA and AD patients than healthy controls. Compared to CAA patients, the Aβ40 level was almost two-fold higher in AD patients (CAA: 8814 pg/mL; AD: 16039 pg/mL; control: 13413 pg/mL; P=0.0014). The CSF level of Aβ42 was higher in CAA patients when compared to the AD group (CAA: 560 pg/mL; AD: 471 pg/mL; control: 789 pg/mL; P=0.034).
Verbeek et al. [7] studied 17 CAA patients (M/F: 11/6), 72 AD patients (M/F: 34/38), and 58 control subjects (M/F: 28/30). This study demonstrates that Aβ40 and Aβ42 levels in CSF are lower in both CAA and AD patients compared to control subjects. Both Aβ40 (CAA: 2912 pg/mL; AD: 3713 pg/mL; control: 4003 pg/mL; P=0.0063, the unpaired t-test was used) and Aβ42 (CAA: 355 pg/mL; AD: 433 pg/mL; control: 838 pg/mL; P=0.039) levels were lower in CAA compared to AD patients. The same result is obtained in 2 other studies included in this review. In a study by Margraf et al. [14], 31 CAA and 28 AD patients with 30 individuals as the control group were studied. When comparing CAA, AD, and controls, they showed that CSF Aβ40 levels did not differ significantly between these groups (CAA: 7008 pg/mL; AD: 7997 pg/mL; control: 8443 pg/mL; P=0.21). They also found that the CSF levels of Aβ42 were lower in both CAA and AD patients compared to the control group (CAA: 347 pg/mL; AD: 340 pg/mL; control: 709 pg/mL; P<0.01). Banerjee et al. [9] assessed 10 CAA, 20 AD, and 10 control subjects. They observed that the Aβ40 level was lower in CAA patients than of AD patients (CAA: 3150 pg/mL; AD: 6470 pg/mL; control: 6890 pg/mL; P<0.001). However, the Aβ42 level was not significantly different between CAA and AD patients and healthy controls, but a slight difference was noticeable when CAA patients were compared to both AD patients and control subjects (CAA: 115 pg/mL; AD: 323 pg/mL; control: 520 pg/mL; P<0.001). Marazuela et al. [17] study of 23 CAA, 26 AD, and 37 controls also reported lowered CSF Aβ40 levels in CAA patients compared to the control group (CAA: 7911 pg/mL; control: 10187 pg/mL) and decreased CSF Aβ42 levels in CAA patients compared to the AD group and to control subjects (CAA: 360 pg/mL; AD: 514.6 pg/mL; control: 895.1 pg/mL). Martínez-Lizana et al. [22] conducted another study on 12 CAA patients (mean age: 69.8 years; 33% males), 42 AD patients (mean age: 67.6 years; 71% males), and 20 control subjects (mean age: 66.5 years; 45% males). The same pattern was observed in this study. They reported decreased CSF levels of Aβ40 and Aβ42 in both CAA and AD patients, compared to the control subject, and lower levels of both of these biomarkers in CAA compared to AD patients (Aβ40; CAA: 5247 pg/mL; AD: 6656 pg/mL; control: 7776 pg/mL, P=0.001), (Aβ42; CAA: 325.75 pg/mL; AD: 336.25 pg/mL; control: 779.5 pg/mL, P=0.001). Also, in another study conducted by Grangeon et al. [19], 63 probable CAA, 27 AD patients, and 21 control patients were studied. They showed that levels of Aβ40 in probable CAA patients were lower than those in AD patients (CAA: 9171.9 pg/mL; AD: 12588 pg/mL; control: 11633 pg/mL; P=0.001, adjusted P=0.001). On the other hand, the values for Aβ42 in CAA patients were higher than in AD patients (CAA: 490.2 pg/mL; AD: 350.7 pg/mL; control: 742.7 pg/mL; P<0.001). In the study by Li et al. [16], Aβ42 concentration is lower in CAA patients than in AD patients, but Aβ40 and p-tau concentrations are higher in CAA compared to AD patients. Based on the studies reviewed, Aβ40 is a better CSF biomarker to differentiate between CAA and AD since its level was reported to be lower in CAA in 9 of 11 articles included in this review.
Contribution of oligomeric tau to CAA and AD
In the study conducted by Li et al. [16], only p-tau was analyzed. The number of CAA cases in this study was too small, decreasing the reliability of the data. Nevertheless, this study reported that the level of p-tau in CSF was higher in CAA compared to AD patients (CAA: 71.8 pg/mL; AD: 47.7 pg/mL). In contrast, Renard et al. [20] found that the CSF p-tau level in AD patients was twice that of CAA patients and control subjects. The level of p-tau was slightly decreased in CAA patients but increased in AD patients compared to control subjects (CAA: 39 pg/mL; AD: 94 pg/mL; control: 41 pg/mL; P<0.0001). CSF t-tau concentration was higher in both CAA and AD patients compared to control subjects with a higher level in the AD group (CAA: 335 pg/mL; AD: 718.5 pg/mL; control: 210 pg/mL; P=0.0031, H-score=8.75). In their previous study, Renard et al. [10] analyzed CSF from 13 CAA, 42 AD patients, and 14 control subjects. They demonstrated that CSF t-tau and p-tau levels increased in AD patients compared to the CAA and control subjects (t-tau, P=0.018; p-tau, P<0.001). The study by Verbeek et al. [7] demonstrates a significant decrease in the p-tau level in the CSF of patients with CAA compared with AD patients (CAA: 66.3 pg/mL; AD: 110 pg/mL; control: 47.9 pg/mL; P=0.0013) and significant increase of its level in the CSF of CAA patients, compared to control subjects (P=0.0009). The CSF t-tau concentrations were significantly lower in CAA subjects than in AD patients (P=0.002). At the same time, there was an increase in both CAA and AD groups when compared to control subjects, and a more than 3-fold increase in the CSF t-tau concentrations was revealed for the AD group compared to controls (P=0.0009, t=3.46; CAA: 395 pg/mL; AD: 712 pg/mL; control: 215 pg/mL). According to the study conducted by Martínez-Lizana et al., t-tau and p-tau levels in CAA and AD patients were also elevated [22]. T-tau CSF levels were increased in both CAA and AD groups compared to healthy controls (P=0.002 for AD patients compared to CAA patients, and P=0.0009 for CAA patients compared to controls). Thus, the t-tau concentration in CAA patients was 2.5 times higher than that in control subjects, which was similar to the 3-fold increase of t-tau level in AD patients compared to control subjects (CAA: 512 pg/mL; AD: 620.8 pg/mL; control: 205 pg/mL). P-tau concentration in the CSF of CAA patients was 69 pg/mL, and in AD patients was 80.3 pg/mL, both of which were approximately 2-fold higher than that of control subjects 37.8 pg/mL (P=0.0013 and t=3.33 comparing CAA and AD groups; P=0.0009). Banerjee et al. [9] reported that the t-tau level was moderately lower in the CSF of patients with CAA compared to the AD group, while the latter had approximately three times higher t-tau level than the control subjects (CAA: 316 pg/mL; AD: 657 pg/mL; control: 250 pg/mL; P<0.001, P=0.35, and P<0.001, respectively, one-way ANOVA followed by Dunn’s post-hoc test). CSF p-tau levels in CAA and AD patients increased compared to control subjects, with the highest level in AD patients (CAA: 62.1 pg/mL; AD: 92.8 pg/mL; control: 49.5 pg/mL). Another study by Renard et al. [10] confirmed lower t-tau and p-tau levels in the CAA group compared to the AD and control groups (for t-tau: CAA: 396 pg/mL; AD: 704 pg/mL; control: 207 pg/mL; P=0.018 and for p-tau; CAA: 45 pg/mL; AD: 92 pg/mL; control: 42 pg/mL; P<0.001). Margraf et al. [14] found that the CSF p-tau level in CAA patients was two-fold higher compared to the control group but decreased in comparison to AD individuals (CAA: 62 pg/mL; AD: 98 pg/mL; control: 32 pg/mL (P<0.01, across all groups, according to the ANOVA). CSF t-tau levels also increased in CAA and AD patients compared to the control group (CAA: 340 pg/mL; AD: 597 pg/mL; control: 211 pg/mL; P<0.01, across all groups). In line with these findings, Renard et al. [20] showed increased CSF t-tau and p-tau levels in AD patients compared to the CAA and control groups. The CSF t-tau values in the AD group were higher than in the CAA group (CAA: 412 pg/mL; AD: 718 pg/mL; control: 210 pg/mL; P<0.01 for comparison across all three groups). The CSF t-tau was higher in CAA compared to controls (P=0.0153), and higher in AD compared to both CAA (P=0.0031) and controls (P<0.001). The same pattern was observed for the CSF p-tau levels (CAA: 46 pg/mL; AD: 94 pg/mL; control: 41 pg/mL; P<0.0001 for all comparisons). However, a study of CSF biomarker concentrations by Marazuella et al. [17] indicates elevated t-tau (CAA: 403 pg/mL; AD: 328.6 pg/mL; control: 231 pg/mL; P=0.002) and p-tau (CAA: 45 pg/mL; AD: 32.4 pg/mL; control: 28 pg/mL; P=0.001) levels in CAA patients compared to the AD and control group. Moreover, in the study by Grangeon et al. [19], CSF levels of t-tau and p-tau are also low in probable CAA patients compared to AD patients (t-tau: CAA: 409.7 pg/mL; AD: 661.9, P=0.006, adjusted P=0.008 and p-tau: CAA: 61.1 pg/mL AD: 92.9 pg/mL; P=0.003, adjusted P=0.004).
Aβ42/Aβ40 ratio in CSF
Two of three studies showed that the Aβ42/Aβ40 ratio in CSF decreases in the AD and CAA groups compared to the normal controls. Banerjee et al. [9] reported that the Aβ42/Aβ40 ratio in CAA patients is lower than in AD patients. However, Grangeon et al. reported that [19] the Aβ42/Aβ40 ratio in CAA patients is higher than in AD patients. Only one study by Li et al. [16] found that this ratio may increase in AD patients compared to CAA patients, but the difference was not significant.
In conclusion, all studies except Li et al. [16] confirmed that p-tau, t-tau, and Aβ40 are reliable biomarkers that may be used in clinical settings to differentiate between CAA and AD. The level of each biomarker in CSF is much higher in AD patients than in CAA patients. However, due to the small sample sizes in the reviewed studies, more research must be conducted to accumulate more reliable data (Figures 2 and 3).

Discussion
CAA is a prevalent cerebrovascular disease that mainly affects elderly patients and results from the deposition of Aβ protein in cortical, subcortical, and leptomeningeal vessels. It is a significant cause of spontaneous ICH and contributes to age-related cognitive decline [3]. Due to the pathological and clinical similarities between this disease and AD, a differential diagnosis based on biochemical markers is required to corroborate the clinical diagnosis. This systematic review investigated Aβ40, Aβ42, p-tau, and t-tau as potential biomarkers allowing differential diagnosis between CAA and AD. According to available studies, the biomarkers in the CSF of AD patients are higher than those in CAA patients [7, 9, 10, 18, 22].
CAA is considered a feature of aging and is strongly related to the pathogenesis of AD [23, 24]. A set of criteria mostly concentrated on radiological imaging called Boston criteria is used to diagnose CAA. According to these criteria, CAA is identified by the occurrence of either two strictly lobar hemorrhagic lesions or a combination of one hemorrhagic lesion and one white matter abnormality. AD is also a significant contributor to ICH in older people. NFTs and the accumulation of Aβ as senile plaques in cerebral gray matter and walls of blood vessels are the main hallmarks in the neuropathological process of AD. These features imply that the failure to prevent the formation of NFTs in neurons [25] and the failure to properly eliminate Aβ from the extracellular compartments of the brain [1, 8] are key contributors to the pathogenesis of AD. These two mechanisms can precede each other, and each of these processes can cause the other, leading to AD pathogenesis [26-28].
Moreover, it has been shown that the accumulation of Aβ can lead to reactive oxygen species (ROS) formation, and oxidative stress can cause higher Aβ production [29]. The majority of AD patients exhibit CAA, which is a characteristic of the aging brain. Dementia was also shown to be associated with severe CAA [30]. According to immunocytochemistry, is mainly found in the cortical artery, capillary walls, and the smaller, distant branches of the leptomeningeal arteries.
Aβ40 was higher in the CSF from AD patients than the control subjects and CAA patients [7, 9, 10, 18, 22] in most studies reviewed. However, Li et al. [16] reported lower levels of Aβ40 in the CSF of AD patients compared to CAA patients due to a drawback in this study. Aβ42 levels were also generally reported to be higher in AD patients than in the CAA and control groups [7, 9, 10, 16, 22]. However, Renard et al. [18] measured lower levels of Aβ42 in AD patients than in CAA patients, which is due to a drawback of this study that the control group had disorders with potentially possible associated alterations in AD biomarkers [31, 32, 33]. On the other hand, the Aβ42 level was similar in AD and patients with CAA in the study by Grangeon et al. [19]. Aβ42 in the CAA and AD groups was lower than controls in most reviewed studies [7, 9, 19-20, 22]. However, in a study by Margraf et al. [14], the Aβ42 levels in CAA patients were higher than in controls. Aβ is known to have a leading part in the pathogenesis of both AD and CAA (Figures 2 and 3).
Naturally, there is a proper balance between producing and clearing Aβ in young human brains, keeping their extracellular level low. This balance might be disturbed in older people and abnormal conditions resulting from impaired Aβ elimination. This condition results in Aβ accumulation in the brain’s parenchyma and the formation of senile plaques, as observed in AD [34]. Senile plaques differ depending on the types of Aβ peptides they contain. The monomeric forms of Aβ peptides consist of 39 to 43 amino acids, with Aβ40 and Aβ42 as the most important forms [35]. Various factors, such as the Aβ40/Aβ42 ratio, determine whether Aβ deposition is parenchymal or vascular. The development of Aβ plaques in CAA and AD differ in how they damage the brain tissue. In AD, the damage is through neuronal and synaptic loss, whereas in CAA, focal lesions are generated in the tissue due to hemorrhage and ischemia in the brain vasculature [36].
Despite Aβ being the most common component of neuritic plaques in AD and vascular amyloid deposits in CAA disease, the length of Aβ peptides appears to vary amongst lesions. Aβ42 is primarily found in neuritic plaques, while Aβ40 peptides are mostly found in the walls of the cortical and leptomeningeal arteries and veins [37-39]. Vascular deposits include Aβ42 as well, but the Aβ40/Aβ42 ratio in vascular deposits is greater than in plaques [40]. The Aβ40/Aβ42 ratio in capillary amyloid deposits, on the other hand, is lower than in arteries and veins and is comparable to neuritic plaques [40, 41].
In most reviewed studies, t-tau and p-tau in the CSF had higher AD levels than controls. High tau has been linked to the production of NFTs and could result from general brain damage. Unlike AD, NFTs are not among the major pathological criteria for CAA [42, 43]. Compared to control subjects, CSF t-tau and p-tau concentrations are higher in CAA patients but lower than levels measured in AD patients [7, 9, 18, 22]. However, Banerjee et al. [9] found no significant elevation in the t-tau and p-tau of CAA patients compared to controls, which may reflect the small sample size in their study. According to Verbeek et al. [7], some AD pathology, even in the non-demented CAA participants, could be a logical explanation for higher tau levels in CAA. They suggested that CSF analysis could help identify CAA patients, although the diagnostic accuracy for distinguishing CAA from AD is still low. CSF study, particularly examining Aβ42 in combination with t-tau, appears to be more effective for distinguishing CAA from non-AD controls [7]. Usually, tau protein plays a role in stabilizing microtubules in neurons, a process required to organize the cytoskeleton. Conformational changes, misfolding, and modifications, such as hyperphosphorylation of tau protein, result in NFT accumulation and disrupt the organization of microtubules in neurodegenerative disorders [44]. The present review considered t-tau and p-tau levels in AD and CAA. Generally, the reviewed articles demonstrated an elevation in both t-tau and p-tau levels in CAA and AD compared to control subjects. Still, in AD patients, the increase was more pronounced [7, 10, 18, 22].
Evidence suggests a significant link between tau pathology and vascular damage and that tau aggregation initiates a vicious cycle that exacerbates vascular damage. Tau aggregation in cerebrovascular diseases has been linked to endothelial dysfunction and cognitive impairment [45, 46]. Several investigations have demonstrated that pathogenic changes in tau in neurons can affect the functions of the brain endothelial cells, disrupting the integrity of microvasculature [47, 48]. For example, vessel wall remodeling of the leptomeningeal arteries has been demonstrated to be an initial, tau pathology-dependent event that may lead to subsequent CAA microvascular pathology in AD patients [48]. Studies on progressive supranuclear palsy patients have identified increased blood-brain-barrier permeability and tau oligomer accumulation in the cerebral microvasculature [49, 50], highlighting the significance of tau aggregates in maintaining the integrity of the cerebral vasculature. You et al. discovered that perivascular tau aggregates near vascular amyloid were linked to reactive astrocytes [44]. These cells are essential for maintaining the blood-brain barrier since their end feet connect directly with vascular endothelial cells [51, 52]. However, in tauopathies linked to CAA, tau has been demonstrated to deposit in astrocyte endfeet [51-53]. Furthermore, patients with chronic traumatic encephalopathy (CTE) have been found to have perivascular tau aggregates [54]. These tau deposits were similarly linked to reactive astrocytes in CTE. Therefore, vascular injury, regardless of the etiology (amyloid buildup or CTE), may initiate an astrocytic response that promotes tau aggregation [55]. Given the importance of tau in various neuropathologies characterized by neurovascular dysfunction and the need for parenchymal amyloid deposition before tau can aggregate, it is plausible to speculate that vascular amyloid would rely on tau to induce neurodegeneration. Also, it could be suggested that partial tau reduction might be a useful method for treating CAA-associated dementias [56] (Figures 2 and 3).
Conclusion
This systematic review compared Aβ and tau protein levels in the CSF of CAA and AD patients to those found in control subjects. Based on this systematic review, CSF p-tau, t-tau, and Aβ40 are not valid biomarkers to distinguish between CAA and AD in the clinical setting, as similar results are not observed. CSF total tau, Aβ42, and phosphorylated tau may be used as biomarkers for AD. Still, as observed in the several studies evaluated, more investigations must be conducted to distinguish between AD and CAA. Moreover, due to the limited sample sizes examined in the evaluated articles, future research is required to obtain more reliable results that can be used to develop novel treatment methods.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Shahid Beheshti University of Medical Sciences (Code: IR.SBMU.RETECH.REC.1399.994).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization and study design: Zahra Razmkhah, Haideh Mosleh, and Komeil Aghazadeh-Habashi; Supervision: Kimia Eyvani; Writing the original draft and final approval: All authors; Review and editing: Mahmood Gorjizad. Zahra Razmkhah and Komeil Aghazadeh-Habashi contributed equally to the paper as the first author.
Conflict of interest
The authors declared no conflict of interest.
References
Alzheimer disease (AD), the most predominant cause of dementia, is characterized by advanced memory impairment and cognitive disabilities. Extracellular amyloid β (Aβ) plaques and hyper-phosphorylated tau in the form of intracellular neurofibrillary tangles (NFTs) are the two main pathological hallmarks of AD [1]. Extracellular Aβ peptides are produced directly by the sequential cleavage of the amyloid precursor protein by β-secretase one and γ-secretase, or they can be transferred into the extracellular space from the endosomes, which contain β- and γ-secretase. Post-translational alterations, including phosphorylation, acetylation, ubiquitination, and truncation, generate pathological tau. These alterations often lead to AD-associated tauopathy, characterized by conformational changes in tau protein, its aggregation, and the formation of toxic oligomers and NFTs [2].
Cerebral amyloid angiopathy (CAA) is a highly prevalent neurological disorder among older people, defined by the accumulation of Aβ peptides within the leptomeninges and walls of small- and medium-size cerebrocortical blood vessels. The deposition of amyloid weakens vasculature and cerebral blood vessel integrity loss, which may manifest in lobar intracerebral hemorrhages (ICH) and cognitive impairment [3]. Aβ peptides participate in the pathogenesis of CAA and are the major components of amyloid plaques found in both cerebral vasculature and brain parenchyma. In vascular deposits, Aβ appears mainly as a 39–40 amino acid peptide (Aβv) in contrast to the 42–43 amino acid peptides that predominate in plaques (Aβp) [4-6]. In CAA, depositions include Aβ40 and Aβ42, with Aβ40 being the dominant form. Increased Aβ40 accumulation is related to the progression of CAA [6-8].
Body fluid biomarkers are typically measured in either cerebrospinal fluid (CSF) or blood due to the ease of collecting these clinical materials. This measure facilitates repeat testing, allowing us to monitor disease-related processes and provide perspectives on disease dynamics [9]. AD biomarkers include CSF levels of total tau (t-tau), phosphorylated tau (p-tau), and Aβ42, which are associated with brain amyloid content and neurodegenerative processes [10, 11]. Changes in Aβ42 concentrations in the CSF (but not the plasma) have been proposed as a possible AD diagnostic criterion [12, 13]. However, increasing evidence indicates that the Aβ42/Aβ40 ratio could benefit the differential diagnosis between AD and other dementias [14]. CSF protein profile in CAA differs from that in AD [10]. The direct evaluation of tau, Aβ peptides, and also other protein markers in the CSF of CAA patients reveals that measurements of t-tau, p-tau181, Aβ40, and Aβ42 could help distinguish CAA and AD patients from controls [14].
Although CAA and AD have numerous molecular similarities, they are not identical [4]. Both are associated with aging, and it has proven difficult to distinguish between vascular brain pathology in CAA and classical AD [15]. CAA can be diagnosed as a standalone disorder or can accompany AD [7]. According to neuropathological studies, more than 90% of AD patients with confirmed plaques and tangles also display signs of CAA, which is caused by Aβ infiltrating into the brain vasculature. However, CAA is also a common condition in non-demented older people, with prevalence rates estimated to range between 20% and 40% [15]. Thus, additional diagnostic criteria are needed to verify the clinical diagnosis due to the pathological and clinical similarities between CAA and AD [16]. Cumulative evidence suggests that CSF biomarkers, t-tau, p-tau, Aβ40, and Aβ42/Aβ40 ratio, can differentiate between CAA and AD [7, 14]. This systematic review aimed to identify all available studies that measured t-tau, p-tau, and or Aβ peptides in the CSF of CAA and AD patients and control subjects. This systematic review aims to establish whether changes in the CSF levels of these proteins could be used as biomarkers, allowing the differential diagnosis between CAA, AD, and control groups. To the best of our knowledge, a systematic review comparing protein biomarkers in the CSF of CAA and AD patients has not been conducted before. Previous systematic reviews have focused on differential diagnosis between AD patients and controls.
Materials and Methods
Literature search strategy
The relevant English articles published from 2009 until 2022 were explored in Medline, Web of Science (WoS), Embase, and Google Scholar. Two authors independently searched using the following key terms and Booleans: ((Alzheimer’s disease) OR (Alzheimer type dementia*) OR (ATD) OR (AD) OR (primary senile degenerative dementia) OR (acute confusional senile dementia) OR (presenile dementia)) AND ((cerebral amyloid angiopathy) OR (CAA) OR (HCHWA) OR (cerebrovascular amyloidosis)) AND ((Aβ40) OR (Aβ42) OR (amyloid-β40) OR (amyloid-β42) OR (tau protein markers) OR (p-tau) OR (t-tau)). The reference lists of all identified studies were also reviewed.
Eligibility criteria
Papers written in English and published in peer-reviewed journals that met all the following inclusion criteria were considered: (1) Observational studies, which (2) report the levels of t-tau, p-tau, Aβ40, Aβ42, or Aβ42/Aβ40 ratio (3) in the CSF of (4) CAA and AD patients, and (5) compare the concentrations of the above proteins in the CSF of CAA and AD patients, or between CAA and or AD patients and a control group. The exclusion criteria were as follows: (1) Non-English studies, (2) animal studies, (3) review articles, conference papers, book chapters, and case reports, (4) studies without randomized sampling, (5) studies reporting the above protein levels only for the AD or CAA patients, (6) studies reporting concentration of the above proteins only in the plasma or the serum of patients, and (7) studies with low-quality scores.
Screening and data extraction
The studies our search assessed were initially screened by their “titles” and “abstracts”. Then, the full texts of selected articles were examined, and eligible articles were included in our study. Relevant data, including authors’ names, year of publication, country of origin, the sample size for the patient and control groups, statistical methods and criteria used, and tau and Aβ concentrations in the CSF, were independently extracted from the included studies by two authors. The results were compared, and any discrepancies were corrected with the help of an additional author.
Results
Study selection
As shown in Figure 1, we extracted 2196 studies for the initial screening after removing duplicated manuscripts.
Diagnostic tools
Most studies selected CAA patients based on the imaging results determined by Boston’s probable CAA criteria. One study [17] used neuropathological confirmation for the CAA diagnosis, and two others [10, 18] employed Chung diagnostic criteria.
While most of the included studies identified AD patients based on the diagnostic criteria of probable AD defined by the National Institute of Neurological and Communicative Disorders and Stroke/AD and Related Disorders Association, Renard et al. [18], Margraf et al. [14], Marazuela et al. [17], and Grangeon et al [19] validated AD diagnosis based on the diagnostic criteria of Alzheimer dementia according to the National Institute on Aging and Alzheimer’s Association.
AD patients had a final diagnosis confirmed in all studies based on imaging data, CSF biomarker measurement, and clinical evaluation. Since CAA leads to lobar hemorrhage, vascular dysfunction, and cognitive decline independent of AD patients with CAA were probably clinically distinctive from AD. However, to prevent overlap and exclude patients with mixed CAA and AD pathology, MRI imaging was performed and reviewed by neuroradiologists for the presence of any cerebral microbleeds, cortical, sub-cortical, and lobar hemorrhagic lesions, and evidence of previous infarction. Specimens from individuals exhibiting these characteristics were excluded from the study. Tau and Aβ levels in the CSF samples were used to identify the probable CAA, AD groups, and healthy individuals. Subjects with evidence or a history of any central nervous system disease, neurodegenerative disease, stroke, arteriovenous malformation, recent brain trauma, or tumor were excluded from the control group.
Contribution of Aβ species to CAA and AD
Ten articles analyzing CSF Aβ40 and Aβ42 levels were included in this review. In the study conducted by Li et al. [16] in China, CSF levels of Aβ40 and Aβ42 in 5 CAA patients were compared to those in 20 AD patients. This study had no control group that could be compared with the CAA and or AD groups. All 5 CAA patients, as well as 12 AD patients, were males. The mean age in the CAA and AD groups was 74.4 and 67.3 years, respectively. The Mean±SD CSF levels of Aβ40 in the CAA group were higher than that in the AD group (CAA: 7111 pg/mL; AD: 5115±2931 pg/mL). However, this difference did not seem statistically significant (P=0.15). Statistical t-test was used to compare the means of Aβ40 levels between the two groups. There was no difference between the concentration of Aβ42 in the CSF from CAA and AD patients (CAA: 660.4 pg/mL; AD: 663.6 pg/mL; P=0.99). In 2016, Renard et al. [10] published a prospective study on 9 patients with CAA-related inflammation in the acute phase. They compared the Aβ protein levels in CSF of these patients to those of 10 CAA patients not displaying acute inflammation, 42 AD patients, 3 patients with primary angiitis of the central nervous system, and 14 controls. Based on the data provided in this study, the CSF concentration of Aβ40 in AD patients was higher than in CAA group and the control subjects. Also, the CAA group was not significantly different from the controls. Aβ42 was higher in CAA (without inflammation) compared to AD patients, though both groups showed a decrease in this biomarker compared to control subjects. In another study, Renard et al. [20] measured CSF biomarkers in 13 CAA patients, 42 AD patients, and 14 controls. They observed that CSF Aβ40 levels in CAA patients decreased significantly compared with the AD patients (CAA: 12138 pg/mL; AD: 16039 pg/mL; P=0.0014). However, the difference between the CSF Aβ40 levels in CAA and control groups was not significant (control: 15424 pg/mL; P=0.20 compared to the CAA group, H-score=1.59). They also observed that CSF Aβ42 levels in the CAA group were significantly lower than in controls (CAA: 560 pg/mL; control: 789 pg/mL; P=0.027). However, the difference between the CAA and the AD group was not significant (AD: 471 pg/mL; P=0.494 compared to the CAA group). Later, in another study, Renard et al. [10] recorded significantly lowered CSF Aβ40 levels (P<0.001) but not significantly lowered Aβ42 levels (P=0.35) in the CAA patient group compared with the AD group. In a prior systematic review and meta-analysis by Charidimou et al. [21], three studies conducted by Renard et al. [18], Verbeek et al. [7], and Martínez-Lizana et al. [22] were investigated. These studies were also included in our work and explained in detail below.
Renard et al. [18] studied 13 CAA patients with lobar hematoma and 42 AD patients compared to 16 control subjects. The CSF levels of Aβ40 and Aβ42 were lower in CAA and AD patients than healthy controls. Compared to CAA patients, the Aβ40 level was almost two-fold higher in AD patients (CAA: 8814 pg/mL; AD: 16039 pg/mL; control: 13413 pg/mL; P=0.0014). The CSF level of Aβ42 was higher in CAA patients when compared to the AD group (CAA: 560 pg/mL; AD: 471 pg/mL; control: 789 pg/mL; P=0.034).
Verbeek et al. [7] studied 17 CAA patients (M/F: 11/6), 72 AD patients (M/F: 34/38), and 58 control subjects (M/F: 28/30). This study demonstrates that Aβ40 and Aβ42 levels in CSF are lower in both CAA and AD patients compared to control subjects. Both Aβ40 (CAA: 2912 pg/mL; AD: 3713 pg/mL; control: 4003 pg/mL; P=0.0063, the unpaired t-test was used) and Aβ42 (CAA: 355 pg/mL; AD: 433 pg/mL; control: 838 pg/mL; P=0.039) levels were lower in CAA compared to AD patients. The same result is obtained in 2 other studies included in this review. In a study by Margraf et al. [14], 31 CAA and 28 AD patients with 30 individuals as the control group were studied. When comparing CAA, AD, and controls, they showed that CSF Aβ40 levels did not differ significantly between these groups (CAA: 7008 pg/mL; AD: 7997 pg/mL; control: 8443 pg/mL; P=0.21). They also found that the CSF levels of Aβ42 were lower in both CAA and AD patients compared to the control group (CAA: 347 pg/mL; AD: 340 pg/mL; control: 709 pg/mL; P<0.01). Banerjee et al. [9] assessed 10 CAA, 20 AD, and 10 control subjects. They observed that the Aβ40 level was lower in CAA patients than of AD patients (CAA: 3150 pg/mL; AD: 6470 pg/mL; control: 6890 pg/mL; P<0.001). However, the Aβ42 level was not significantly different between CAA and AD patients and healthy controls, but a slight difference was noticeable when CAA patients were compared to both AD patients and control subjects (CAA: 115 pg/mL; AD: 323 pg/mL; control: 520 pg/mL; P<0.001). Marazuela et al. [17] study of 23 CAA, 26 AD, and 37 controls also reported lowered CSF Aβ40 levels in CAA patients compared to the control group (CAA: 7911 pg/mL; control: 10187 pg/mL) and decreased CSF Aβ42 levels in CAA patients compared to the AD group and to control subjects (CAA: 360 pg/mL; AD: 514.6 pg/mL; control: 895.1 pg/mL). Martínez-Lizana et al. [22] conducted another study on 12 CAA patients (mean age: 69.8 years; 33% males), 42 AD patients (mean age: 67.6 years; 71% males), and 20 control subjects (mean age: 66.5 years; 45% males). The same pattern was observed in this study. They reported decreased CSF levels of Aβ40 and Aβ42 in both CAA and AD patients, compared to the control subject, and lower levels of both of these biomarkers in CAA compared to AD patients (Aβ40; CAA: 5247 pg/mL; AD: 6656 pg/mL; control: 7776 pg/mL, P=0.001), (Aβ42; CAA: 325.75 pg/mL; AD: 336.25 pg/mL; control: 779.5 pg/mL, P=0.001). Also, in another study conducted by Grangeon et al. [19], 63 probable CAA, 27 AD patients, and 21 control patients were studied. They showed that levels of Aβ40 in probable CAA patients were lower than those in AD patients (CAA: 9171.9 pg/mL; AD: 12588 pg/mL; control: 11633 pg/mL; P=0.001, adjusted P=0.001). On the other hand, the values for Aβ42 in CAA patients were higher than in AD patients (CAA: 490.2 pg/mL; AD: 350.7 pg/mL; control: 742.7 pg/mL; P<0.001). In the study by Li et al. [16], Aβ42 concentration is lower in CAA patients than in AD patients, but Aβ40 and p-tau concentrations are higher in CAA compared to AD patients. Based on the studies reviewed, Aβ40 is a better CSF biomarker to differentiate between CAA and AD since its level was reported to be lower in CAA in 9 of 11 articles included in this review.
Contribution of oligomeric tau to CAA and AD
In the study conducted by Li et al. [16], only p-tau was analyzed. The number of CAA cases in this study was too small, decreasing the reliability of the data. Nevertheless, this study reported that the level of p-tau in CSF was higher in CAA compared to AD patients (CAA: 71.8 pg/mL; AD: 47.7 pg/mL). In contrast, Renard et al. [20] found that the CSF p-tau level in AD patients was twice that of CAA patients and control subjects. The level of p-tau was slightly decreased in CAA patients but increased in AD patients compared to control subjects (CAA: 39 pg/mL; AD: 94 pg/mL; control: 41 pg/mL; P<0.0001). CSF t-tau concentration was higher in both CAA and AD patients compared to control subjects with a higher level in the AD group (CAA: 335 pg/mL; AD: 718.5 pg/mL; control: 210 pg/mL; P=0.0031, H-score=8.75). In their previous study, Renard et al. [10] analyzed CSF from 13 CAA, 42 AD patients, and 14 control subjects. They demonstrated that CSF t-tau and p-tau levels increased in AD patients compared to the CAA and control subjects (t-tau, P=0.018; p-tau, P<0.001). The study by Verbeek et al. [7] demonstrates a significant decrease in the p-tau level in the CSF of patients with CAA compared with AD patients (CAA: 66.3 pg/mL; AD: 110 pg/mL; control: 47.9 pg/mL; P=0.0013) and significant increase of its level in the CSF of CAA patients, compared to control subjects (P=0.0009). The CSF t-tau concentrations were significantly lower in CAA subjects than in AD patients (P=0.002). At the same time, there was an increase in both CAA and AD groups when compared to control subjects, and a more than 3-fold increase in the CSF t-tau concentrations was revealed for the AD group compared to controls (P=0.0009, t=3.46; CAA: 395 pg/mL; AD: 712 pg/mL; control: 215 pg/mL). According to the study conducted by Martínez-Lizana et al., t-tau and p-tau levels in CAA and AD patients were also elevated [22]. T-tau CSF levels were increased in both CAA and AD groups compared to healthy controls (P=0.002 for AD patients compared to CAA patients, and P=0.0009 for CAA patients compared to controls). Thus, the t-tau concentration in CAA patients was 2.5 times higher than that in control subjects, which was similar to the 3-fold increase of t-tau level in AD patients compared to control subjects (CAA: 512 pg/mL; AD: 620.8 pg/mL; control: 205 pg/mL). P-tau concentration in the CSF of CAA patients was 69 pg/mL, and in AD patients was 80.3 pg/mL, both of which were approximately 2-fold higher than that of control subjects 37.8 pg/mL (P=0.0013 and t=3.33 comparing CAA and AD groups; P=0.0009). Banerjee et al. [9] reported that the t-tau level was moderately lower in the CSF of patients with CAA compared to the AD group, while the latter had approximately three times higher t-tau level than the control subjects (CAA: 316 pg/mL; AD: 657 pg/mL; control: 250 pg/mL; P<0.001, P=0.35, and P<0.001, respectively, one-way ANOVA followed by Dunn’s post-hoc test). CSF p-tau levels in CAA and AD patients increased compared to control subjects, with the highest level in AD patients (CAA: 62.1 pg/mL; AD: 92.8 pg/mL; control: 49.5 pg/mL). Another study by Renard et al. [10] confirmed lower t-tau and p-tau levels in the CAA group compared to the AD and control groups (for t-tau: CAA: 396 pg/mL; AD: 704 pg/mL; control: 207 pg/mL; P=0.018 and for p-tau; CAA: 45 pg/mL; AD: 92 pg/mL; control: 42 pg/mL; P<0.001). Margraf et al. [14] found that the CSF p-tau level in CAA patients was two-fold higher compared to the control group but decreased in comparison to AD individuals (CAA: 62 pg/mL; AD: 98 pg/mL; control: 32 pg/mL (P<0.01, across all groups, according to the ANOVA). CSF t-tau levels also increased in CAA and AD patients compared to the control group (CAA: 340 pg/mL; AD: 597 pg/mL; control: 211 pg/mL; P<0.01, across all groups). In line with these findings, Renard et al. [20] showed increased CSF t-tau and p-tau levels in AD patients compared to the CAA and control groups. The CSF t-tau values in the AD group were higher than in the CAA group (CAA: 412 pg/mL; AD: 718 pg/mL; control: 210 pg/mL; P<0.01 for comparison across all three groups). The CSF t-tau was higher in CAA compared to controls (P=0.0153), and higher in AD compared to both CAA (P=0.0031) and controls (P<0.001). The same pattern was observed for the CSF p-tau levels (CAA: 46 pg/mL; AD: 94 pg/mL; control: 41 pg/mL; P<0.0001 for all comparisons). However, a study of CSF biomarker concentrations by Marazuella et al. [17] indicates elevated t-tau (CAA: 403 pg/mL; AD: 328.6 pg/mL; control: 231 pg/mL; P=0.002) and p-tau (CAA: 45 pg/mL; AD: 32.4 pg/mL; control: 28 pg/mL; P=0.001) levels in CAA patients compared to the AD and control group. Moreover, in the study by Grangeon et al. [19], CSF levels of t-tau and p-tau are also low in probable CAA patients compared to AD patients (t-tau: CAA: 409.7 pg/mL; AD: 661.9, P=0.006, adjusted P=0.008 and p-tau: CAA: 61.1 pg/mL AD: 92.9 pg/mL; P=0.003, adjusted P=0.004).
Aβ42/Aβ40 ratio in CSF
Two of three studies showed that the Aβ42/Aβ40 ratio in CSF decreases in the AD and CAA groups compared to the normal controls. Banerjee et al. [9] reported that the Aβ42/Aβ40 ratio in CAA patients is lower than in AD patients. However, Grangeon et al. reported that [19] the Aβ42/Aβ40 ratio in CAA patients is higher than in AD patients. Only one study by Li et al. [16] found that this ratio may increase in AD patients compared to CAA patients, but the difference was not significant.
In conclusion, all studies except Li et al. [16] confirmed that p-tau, t-tau, and Aβ40 are reliable biomarkers that may be used in clinical settings to differentiate between CAA and AD. The level of each biomarker in CSF is much higher in AD patients than in CAA patients. However, due to the small sample sizes in the reviewed studies, more research must be conducted to accumulate more reliable data (Figures 2 and 3).
Discussion
CAA is a prevalent cerebrovascular disease that mainly affects elderly patients and results from the deposition of Aβ protein in cortical, subcortical, and leptomeningeal vessels. It is a significant cause of spontaneous ICH and contributes to age-related cognitive decline [3]. Due to the pathological and clinical similarities between this disease and AD, a differential diagnosis based on biochemical markers is required to corroborate the clinical diagnosis. This systematic review investigated Aβ40, Aβ42, p-tau, and t-tau as potential biomarkers allowing differential diagnosis between CAA and AD. According to available studies, the biomarkers in the CSF of AD patients are higher than those in CAA patients [7, 9, 10, 18, 22].
CAA is considered a feature of aging and is strongly related to the pathogenesis of AD [23, 24]. A set of criteria mostly concentrated on radiological imaging called Boston criteria is used to diagnose CAA. According to these criteria, CAA is identified by the occurrence of either two strictly lobar hemorrhagic lesions or a combination of one hemorrhagic lesion and one white matter abnormality. AD is also a significant contributor to ICH in older people. NFTs and the accumulation of Aβ as senile plaques in cerebral gray matter and walls of blood vessels are the main hallmarks in the neuropathological process of AD. These features imply that the failure to prevent the formation of NFTs in neurons [25] and the failure to properly eliminate Aβ from the extracellular compartments of the brain [1, 8] are key contributors to the pathogenesis of AD. These two mechanisms can precede each other, and each of these processes can cause the other, leading to AD pathogenesis [26-28].
Moreover, it has been shown that the accumulation of Aβ can lead to reactive oxygen species (ROS) formation, and oxidative stress can cause higher Aβ production [29]. The majority of AD patients exhibit CAA, which is a characteristic of the aging brain. Dementia was also shown to be associated with severe CAA [30]. According to immunocytochemistry, is mainly found in the cortical artery, capillary walls, and the smaller, distant branches of the leptomeningeal arteries.
Aβ40 was higher in the CSF from AD patients than the control subjects and CAA patients [7, 9, 10, 18, 22] in most studies reviewed. However, Li et al. [16] reported lower levels of Aβ40 in the CSF of AD patients compared to CAA patients due to a drawback in this study. Aβ42 levels were also generally reported to be higher in AD patients than in the CAA and control groups [7, 9, 10, 16, 22]. However, Renard et al. [18] measured lower levels of Aβ42 in AD patients than in CAA patients, which is due to a drawback of this study that the control group had disorders with potentially possible associated alterations in AD biomarkers [31, 32, 33]. On the other hand, the Aβ42 level was similar in AD and patients with CAA in the study by Grangeon et al. [19]. Aβ42 in the CAA and AD groups was lower than controls in most reviewed studies [7, 9, 19-20, 22]. However, in a study by Margraf et al. [14], the Aβ42 levels in CAA patients were higher than in controls. Aβ is known to have a leading part in the pathogenesis of both AD and CAA (Figures 2 and 3).
Naturally, there is a proper balance between producing and clearing Aβ in young human brains, keeping their extracellular level low. This balance might be disturbed in older people and abnormal conditions resulting from impaired Aβ elimination. This condition results in Aβ accumulation in the brain’s parenchyma and the formation of senile plaques, as observed in AD [34]. Senile plaques differ depending on the types of Aβ peptides they contain. The monomeric forms of Aβ peptides consist of 39 to 43 amino acids, with Aβ40 and Aβ42 as the most important forms [35]. Various factors, such as the Aβ40/Aβ42 ratio, determine whether Aβ deposition is parenchymal or vascular. The development of Aβ plaques in CAA and AD differ in how they damage the brain tissue. In AD, the damage is through neuronal and synaptic loss, whereas in CAA, focal lesions are generated in the tissue due to hemorrhage and ischemia in the brain vasculature [36].
Despite Aβ being the most common component of neuritic plaques in AD and vascular amyloid deposits in CAA disease, the length of Aβ peptides appears to vary amongst lesions. Aβ42 is primarily found in neuritic plaques, while Aβ40 peptides are mostly found in the walls of the cortical and leptomeningeal arteries and veins [37-39]. Vascular deposits include Aβ42 as well, but the Aβ40/Aβ42 ratio in vascular deposits is greater than in plaques [40]. The Aβ40/Aβ42 ratio in capillary amyloid deposits, on the other hand, is lower than in arteries and veins and is comparable to neuritic plaques [40, 41].
In most reviewed studies, t-tau and p-tau in the CSF had higher AD levels than controls. High tau has been linked to the production of NFTs and could result from general brain damage. Unlike AD, NFTs are not among the major pathological criteria for CAA [42, 43]. Compared to control subjects, CSF t-tau and p-tau concentrations are higher in CAA patients but lower than levels measured in AD patients [7, 9, 18, 22]. However, Banerjee et al. [9] found no significant elevation in the t-tau and p-tau of CAA patients compared to controls, which may reflect the small sample size in their study. According to Verbeek et al. [7], some AD pathology, even in the non-demented CAA participants, could be a logical explanation for higher tau levels in CAA. They suggested that CSF analysis could help identify CAA patients, although the diagnostic accuracy for distinguishing CAA from AD is still low. CSF study, particularly examining Aβ42 in combination with t-tau, appears to be more effective for distinguishing CAA from non-AD controls [7]. Usually, tau protein plays a role in stabilizing microtubules in neurons, a process required to organize the cytoskeleton. Conformational changes, misfolding, and modifications, such as hyperphosphorylation of tau protein, result in NFT accumulation and disrupt the organization of microtubules in neurodegenerative disorders [44]. The present review considered t-tau and p-tau levels in AD and CAA. Generally, the reviewed articles demonstrated an elevation in both t-tau and p-tau levels in CAA and AD compared to control subjects. Still, in AD patients, the increase was more pronounced [7, 10, 18, 22].
Evidence suggests a significant link between tau pathology and vascular damage and that tau aggregation initiates a vicious cycle that exacerbates vascular damage. Tau aggregation in cerebrovascular diseases has been linked to endothelial dysfunction and cognitive impairment [45, 46]. Several investigations have demonstrated that pathogenic changes in tau in neurons can affect the functions of the brain endothelial cells, disrupting the integrity of microvasculature [47, 48]. For example, vessel wall remodeling of the leptomeningeal arteries has been demonstrated to be an initial, tau pathology-dependent event that may lead to subsequent CAA microvascular pathology in AD patients [48]. Studies on progressive supranuclear palsy patients have identified increased blood-brain-barrier permeability and tau oligomer accumulation in the cerebral microvasculature [49, 50], highlighting the significance of tau aggregates in maintaining the integrity of the cerebral vasculature. You et al. discovered that perivascular tau aggregates near vascular amyloid were linked to reactive astrocytes [44]. These cells are essential for maintaining the blood-brain barrier since their end feet connect directly with vascular endothelial cells [51, 52]. However, in tauopathies linked to CAA, tau has been demonstrated to deposit in astrocyte endfeet [51-53]. Furthermore, patients with chronic traumatic encephalopathy (CTE) have been found to have perivascular tau aggregates [54]. These tau deposits were similarly linked to reactive astrocytes in CTE. Therefore, vascular injury, regardless of the etiology (amyloid buildup or CTE), may initiate an astrocytic response that promotes tau aggregation [55]. Given the importance of tau in various neuropathologies characterized by neurovascular dysfunction and the need for parenchymal amyloid deposition before tau can aggregate, it is plausible to speculate that vascular amyloid would rely on tau to induce neurodegeneration. Also, it could be suggested that partial tau reduction might be a useful method for treating CAA-associated dementias [56] (Figures 2 and 3).
Conclusion
This systematic review compared Aβ and tau protein levels in the CSF of CAA and AD patients to those found in control subjects. Based on this systematic review, CSF p-tau, t-tau, and Aβ40 are not valid biomarkers to distinguish between CAA and AD in the clinical setting, as similar results are not observed. CSF total tau, Aβ42, and phosphorylated tau may be used as biomarkers for AD. Still, as observed in the several studies evaluated, more investigations must be conducted to distinguish between AD and CAA. Moreover, due to the limited sample sizes examined in the evaluated articles, future research is required to obtain more reliable results that can be used to develop novel treatment methods.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Shahid Beheshti University of Medical Sciences (Code: IR.SBMU.RETECH.REC.1399.994).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors contributions
Conceptualization and study design: Zahra Razmkhah, Haideh Mosleh, and Komeil Aghazadeh-Habashi; Supervision: Kimia Eyvani; Writing the original draft and final approval: All authors; Review and editing: Mahmood Gorjizad. Zahra Razmkhah and Komeil Aghazadeh-Habashi contributed equally to the paper as the first author.
Conflict of interest
The authors declared no conflict of interest.
References
- Ittner LM, Götz J. Amyloid-β and tau-a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011; 12(2):65-72. [DOI:10.1038/nrn2967] [PMID]
- Chang HY, Sang TK, Chiang AS. Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. J Biomed Sci. 2018; 25(1):54. [DOI:10.1186/s12929-018-0457-x] [PMID]
- Yamada M. Cerebral amyloid angiopathy: Emerging concepts. J Stroke. 2015; 17(1):17-30. [DOI:10.5853/jos.2015.17.1.17] [PMID]
- Greenberg SM, Cho HS, O'Donnell HC, Rosand J, Segal AZ, Younkin LH, et al. Plasma beta-amyloid peptide, transforming growth factor-beta 1, and risk for cerebral amyloid angiopathy. Ann N Y Acad Sci. 2000; 903:144-9. [DOI:10.1111/j.1749-6632.2000.tb06361.x] [PMID]
- Mann DM, Iwatsubo T, Ihara Y, Cairns NJ, Lantos PL, Bogdanovic N, et al. Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol. 1996; 148(4):1257-66. [PMID]
- Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: Accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998; 57(4):353-9. [DOI:10.1097/00005072-199804000-00008] [PMID]
- Verbeek MM, Kremer BP, Rikkert MO, Van Domburg PH, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid β40 is decreased in cerebral amyloid angiopathy. Ann Neurol. 2009; 66(2):245-9. [DOI:10.1002/ana.21694] [PMID]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron. 1994; 13(1):45-53. [DOI:10.1016/0896-6273(94)90458-8] [PMID]
- Banerjee G, Ambler G, Keshavan A, Paterson RW, Foiani MS, Toombs J, et al., Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy. J Alzheimers Dis. 2020; 74(4):1189-201. [DOI:10.3233/JAD-191254] [PMID]
- Renard D, Wacongne A, Ayrignac X, Charif M, Fourcade G, Azakri S, et al. Cerebrospinal Fluid Alzheimer’s Disease Biomarkers in cerebral amyloid angiopathy-related inflammation. J Alzheimers Dis. 2016; 50(3):759-64.[DOI:10.3233/JAD-150621] [PMID]
- Gabelle A, Dumurgier J, Vercruysse O, Paquet C, Bombois S, Laplanche JL, et al. Impact of the 2008-2012 French Alzheimer Plan on the use of cerebrospinal fluid biomarkers in research memory center: The PLM Study. J Alzheimers Dis. 2013; 34(1):297-305. [DOI:10.3233/JAD-121549] [PMID]
- Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, et al., Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995; 38(4):643-8. [DOI:10.1002/ana.410380413] [PMID]
- Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, et al. Cerebrospinal Fluid β-Amyloid(1-42) in Alzheimer Disease: Differences between early- and late-onset alzheimer disease and stability during the course of disease. Arch Neurol. 1999; 56(6):673-80. [DOI:10.1001/archneur.56.6.673] [PMID]
- Margraf NG, Jensen-Kondering U, Weiler C, Leypoldt F, Maetzler W, Philippen S, et al. Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy: New data and quantitative meta-analysis. Front Aging Neurosci. 2022; 14:783996. [DOI:10.3389/fnagi.2022.783996] [PMID]
- Janson ChG. AD and CAA: Independent risk factors for dementia. Sci Transl Med. 2015; 7(318):318ec214. [DOI:10.1126/scitranslmed.aad9005]
- Li YF, Ge FF, Zhang Y, You H, Zhang ZX. Cerebrospinal fluid biomarkers in dementia patients with cerebral amyloid angiopathy. Chin Med Sci J. 2015; 30(3):170-3. [DOI:10.1016/S1001-9294(15)30042-0] [PMID]
- Marazuela P, Solé M, Bonaterra-Pastra A, Pizarro J, Camacho J, Martínez-Sáez E, et al. MFG-E8 (LACTADHERIN): A novel marker associated with cerebral amyloid angiopathy. Acta Neuropathol Commun. 2021; 9(1):1-17. [DOI:10.1186/s40478-021-01257-9]
- Renard D, Gabelle A, Hirtz C, Demattei C, Thouvenot E, Lehmann S. Cerebrospinal fluid Alzheimer’s disease biomarkers in isolated supratentorial cortical superficial siderosis. J Alzheimers Dis. 2016. 54(4):1291-5. [DOI:10.3233/JAD-160400] [PMID]
- Grangeon L, Paquet C, Guey S, Zarea A, Martinaud O, Rotharmel M,et al. Cerebrospinal fluid profile of tau, phosphorylated tau, Aβ 42, and Aβ 40 in probable cerebral amyloid angiopathy. J Alzheimers Dis. 2022; 87(2):791-802. [DOI:10.3233/JAD-215208] [PMID]
- Renard D, Castelnovo G, Wacongne A, Le Floch A, Thouvenot E, Mas J, et al. Interest of CSF biomarker analysis in possible cerebral amyloid angiopathy cases defined by the modified Boston criteria. J Neurol. 2012; 259(11):2429-33. [DOI:10.1007/s00415-012-6520-8] [PMID]
- Charidimou A, Friedrich JO, Greenberg SM, Viswanathan A. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: A meta-analysis. Neurology. 2018; 90(9):e754-62. [DOI:10.1212/WNL.0000000000005030] [PMID]
- Martínez-Lizana E, Carmona-Iragui M, Alcolea D, Gómez-Choco M, Vilaplana E, Sánchez-Saudinós MB, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: An ARIA before the tsunami. J Cereb Blood Flow Metab. 2015; 35(5):710-7. [DOI:10.1038/jcbfm.2015.25] [PMID]
- Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer’s disease. Pro-CAA position statement. Neurobiol Aging. 2004; 25(5):589-97; discussion 603-4. [DOI:10.1016/j.neurobiolaging.2004.02.003] [PMID]
- Weller RO, Nicoll JA. Cerebral amyloid angiopathy: Pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res. 2003; 25(6):611-6. [DOI:10.1179/016164103101202057] [PMID]
- Layfield R, Alban A, Mayer RJ, Lowe J. The ubiquitin protein catabolic disorders. Neuropathol Appl Neurobiol. 2001; 27(3):171-9. [DOI:10.1046/j.1365-2990.2001.00335.x] [PMID]
- Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: Defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol Appl Neurobiol. 2003; 29(2):106-17. [DOI:10.1046/j.1365-2990.2003.00424.x] [PMID]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: Amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998; 153(3):725-33. [DOI:10.1016/S0002-9440(10)65616-7] [PMID]
- Dibaj M, Haghi M, Safaralizadeh R, Saberi A. The role of EZH2 and its regulatory lncRNAs as a serum-based biomarker in Alzheimer’s disease. Mol Biol Rep. 2024; 51(1):866. [DOI:10.1007/s11033-024-09802-0] [PMID]
- Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C. Monge-bonilla, molecular pathogenesis of Alzheimer’s disease: An update. Ann Neurosci. 2017; 24(1):46-54. [DOI:10.1159/000464422] [PMID]
- Neuropathology Group, Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet. 2001; 357(9251):169-75. [DOI: 10.1016/s0140-6736(00)03589-3] [PMID]
- Laiterä T, Kurki MI, Pursiheimo JP, Zetterberg H, Helisalmi S, Rauramaa T, et al. The expression of transthyretin and amyloid-β protein precursor is altered in the brain of idiopathic normal pressure hydrocephalus patients. J Alzheimers Dis. 2015; 48(4):959-68. [DOI:10.3233/JAD-150268] [PMID]
- Jingami N, Asada-Utsugi M, Uemura K, Noto R, Takahashi M, Ozaki A, et al. Idiopathic normal pressure hydrocephalus has a different cerebrospinal fluid biomarker profile from Alzheimer’s disease. J Alzheimers Dis. 2015; 45(1):109-15. [DOI:10.3233/JAD-142622] [PMID]
- Herukka SK, Rummukainen J, Ihalainen J, von Und Zu Fraunberg M, Koivisto AM, et al. Amyloid-β and Tau Dynamics in human brain interstitial fluid in patients with suspected normal pressure hydrocephalus. J Alzheimers Dis. 2015; 46(1):261-9. [DOI:10.3233/JAD-142862] [PMID]
- Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med Princ Pract. 2015; 24(1):1-10. [DOI:10.1159/000369101] [PMID]
- Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid β protein deposits in the aged brain. Sci Aging Knowledge Environ. 2006; 2006(6):re1. [DOI:10.1126/sageke.2006.6.re1] [PMID]
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020; 16(1):30-42. [DOI:10.1038/s41582-019-0281-2] [PMID]
- Gravina SA, Ho L, Eckman CB, Long KE, Otvos L Jr, Younkin LH, Suzuki N, Younkin SG. Amyloid beta protein (A beta) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem. 1995; 270(13):7013-6. [DOI: 10.1074/jbc.270.13.7013] [PMID]
- Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer′ s disease. Arch Biochem Biophys. 1993; 301(1):41-52. [DOI:10.1006/abbi.1993.1112] [PMID]
- Kakuda N, Miyasaka T, Iwasaki N, Nirasawa T, Wada-Kakuda S, Takahashi-Fujigasaki J, et al. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol Commun. 2017; 5(1):73.[DOI:10.1186/s40478-017-0477-x] [PMID]
- Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, et al. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A. 1993; 90(22):10836-40. [DOI:10.1073/pnas.90.22.10836] [PMID]
- Attems J, Lintner F, Jellinger KA. Amyloid β peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol. 2004; 107(4):283-91. [DOI:10.1007/s00401-004-0822-6] [PMID]
- Maat-Schieman ML, Radder CM, van Duinen SG, Haan J, Roos RA. Hereditary cerebral hemorrhage with amyloidosis (Dutch): A model for congophilic plaque formation without neurofibrillary pathology. Acta Neuropathol. 1994; 88(4):371-8. [DOI:10.1007/BF00310382] [PMID]
- Mandybur TI. Cerebral amyloid angiopathy: The vascular pathology and complications. J Neuropathol Exp Neurol. 1986; 45(1):79-90. [DOI:10.1097/00005072-198601000-00007]
- You Y, Perkins A, Cisternas P, Muñoz B, Taylor X, You Y, et al. Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol Commun. 2019; 7(1):26. [DOI:10.1186/s40478-019-0680-z] [PMID]
- Kim HJ, Park S, Cho H, Jang YK, San Lee J, Jang H, et al. Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18F-AV1451 positron emission tomography imaging. JAMA Neurol. 2018; 75(8):999-1007.[DOI:10.1001/jamaneurol.2018.0975] [PMID]
- Nation DA, Edmonds EC, Bangen KJ, Delano-Wood L, Scanlon BK, Han SD, et al. Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol. 2015; 72(5):546-53. [DOI:10.1001/jamaneurol.2014.4477] [PMID]
- Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, et al. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci U S A. 2018; 115(6):E1289-98. [DOI:10.1073/pnas.1710329115] [PMID]
- Merlini M, Wanner D, Nitsch RM. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol. 2016; 131(5):737-52. [DOI:10.1007/s00401-016-1560-2] [PMID]
- Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, et al. Cerebral microvascular accumulation of tau oligomers in Alzheimer’s disease and related tauopathies. Aging Dis. 2017; 8(3):257-66. [DOI:10.14336/AD.2017.0112] [PMID]
- Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm (Vienna). 2008; 115(7):1001-9. [DOI:10.1007/s00702-008-0030-y] [PMID]
- Komori T. Tau-positive dial Inclusions in Progressive Supranuclear Palsy, Corticobasal Degeneration and Pick’s disease. Brain Pathol. 1999; 9(4):663-79. [DOI:10.1111/j.1750-3639.1999.tb00549.x] [PMID]
- Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last). Trends Neurosci. 2003; 26(10):520-2. [DOI:10.1016/j.tins.2003.08.006]
- Vidal R, Calero M, Piccardo P, Farlow MR, Unverzagt FW, Méndez E, et al. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol. 2000; 100(1):1-12. [DOI:10.1007/s004010051186] [PMID]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. 2017; 318(4):360-70. [DOI:10.1001/jama.2017.8334] [PMID]
- Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2016; 75(1):19-34. [DOI:10.1093/jnen/nlv001] [PMID]
- Cisternas P, Taylor X, Lasagna-Reeves CA. The amyloid-tau-neuroinflammation axis in the context of cerebral amyloid angiopathy. Int J Mol Sci. 2019; 20(24):6319. [DOI:10.3390/ijms20246319] [PMID]
Type of Study: Review |
Subject:
Special
Received: 2024/04/23 | Accepted: 2024/10/5 | Published: 2024/10/5
Received: 2024/04/23 | Accepted: 2024/10/5 | Published: 2024/10/5
Send email to the article author
| Rights and permissions | |
 | This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
