Mon, Feb 16, 2026
Volume 7, Issue 4 (Autumn 2021)
Caspian J Neurol Sci 2021, 7(4): 236-243 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Koohmanaee S, Tamimi A, Ahmadimacciani S, Tamimi A, Aminzadeh V, Zarkesh M, et al . The Coexistence of Gonadal Dysgenesis With Mayer-rokitansky-küster-hauser Syndrome, and Dandy-Walker Variant. Caspian J Neurol Sci 2021; 7 (4) :236-243
URL: http://cjns.gums.ac.ir/article-1-470-en.html
URL: http://cjns.gums.ac.ir/article-1-470-en.html
Shahin Koohmanaee1 

 , Amirhossein Tamimi2 , Soroush Ahmadimacciani2 , Atena Tamimi3 , Vahid Aminzadeh1 , Marjaneh Zarkesh1 , Seyyedeh Azadeh Hoseini Nouri1 , Fatemeh Rajaeipoor1 , Manijeh Tabrizi1 , Setila Dalili *4
, Amirhossein Tamimi2 , Soroush Ahmadimacciani2 , Atena Tamimi3 , Vahid Aminzadeh1 , Marjaneh Zarkesh1 , Seyyedeh Azadeh Hoseini Nouri1 , Fatemeh Rajaeipoor1 , Manijeh Tabrizi1 , Setila Dalili *4
, Amirhossein Tamimi2 , Soroush Ahmadimacciani2 , Atena Tamimi3 , Vahid Aminzadeh1 , Marjaneh Zarkesh1 , Seyyedeh Azadeh Hoseini Nouri1 , Fatemeh Rajaeipoor1 , Manijeh Tabrizi1 , Setila Dalili *4
1- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran.
2- Student Research Committee, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran.
3- Student Research Committee, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
4- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran. ,setiladalili1346@yahoo.com
2- Student Research Committee, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran.
3- Student Research Committee, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
4- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran. ,
Full-Text [PDF 1347 kb]
(788 Downloads)
| Abstract (HTML) (2965 Views)
Full-Text: (1138 Views)
Introduction
Gonadal dysgenesis is depicted by the absent or underdeveloped ovaries in females with primary amenorrhea and leads to variable degrees of hypogonadism and impuberism [1]. Patients with gonadal dysgenesis can have different karyotypes in the forms of 46 XX, 45 XO, mosaicism, or deletion of a specific part of the X chromosome [2].
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, the second most common cause of primary amenorrhea following gonadal dysgenesis, is related to the congenital absence or hypoplasia of the uterus and upper two-thirds of the vagina in a female with normal phenotype and karyotype. It affects nearly 1 out of 5,000 newborn girls [3]. MRKH syndrome can be whether isolated (type 1) or associated with renal and skeletal malformations, auditory defects, and occasionally, anomalies of the heart and digits (type 2) [4].
Dandy Walker Complex (DWC) is a Central Nervous System (CNS) malformation, which comprises of four types: 1- Dandy-Walker malformation marked by a triad of agenesis or hypoplasia of the cerebellar vermis, cystic dilatation of the fourth ventricle, and an enlarged posterior fossa; 2- Dandy-Walker variant associated with vermis hypoplasia, cystic dilatation of the fourth ventricle, and normal posterior fossa; 3- Mega cisternae Magna delineated by normal vermis and fourth ventricle but large posterior fossa; and 4- Posterior fossa arachnoid cyst [5].
Although the coexistence of gonadal dysgenesis and MRKH has been reported, it is still quite infrequent. To the extent that authors searched, just one study reported the association between Rokitansky sequence and Dandy-Walker malformation [6]. Here, the authors aimed to report a case with gonadal dysgenesis, MRKH, and the Dandy-Walker variant altogether.
Case Presentation
A 15-year-old girl was presented with primary amenorrhea. She had been experiencing two-day-lasting episodes of fatigue, lethargy, abdominal pain, and monthly vaginal discharge for two years.
Her birth weight was 2400 grams. She underwent intestinal surgery at two days of age because of excessive vomiting due to duodenal atresia. She was also hospitalized due to asthma and allergy four years before the recent attendance at this clinic. Moreover, due to blurred vision and repetitive drop attacks remaining for several months, brain Magnetic Resonance Imaging (MRI) was performed and revealed the Dandy-Walker variant. Subsequently, she was on piracetam, folic acid, and vitamin E for six months, which was followed by the resolution of symptoms and cessation of treatment four years before admission. The patient was the firstborn of first-degree cousin marriage. She was born preterm by Natural Vaginal Delivery (NVD). The mother had been pregnant five times, three of which led to NVD, and she had had two uninduced abortions.
On examination, the patient was in thelarche stage three and pubarche stage two. Abdominal obesity and striae were identified. Axillary hair was very sparse. She was 151 cm in height and 65 kg in weight with a Body Mass Index (BMI) of 28.5 (percentile= 95th). Her father’s and mother’s heights were 177 and 155 cm, respectively.
On pelvic transabdominal ultrasound, the uterus was not seen and both ovaries looked atrophic, small, and underdeveloped with no follicles, but the vagina was normal. The cervix and uterus could not be found. There was no evidence of free fluid in the pelvic cavity during both sonographies. Other organs seemed normal on ultrasound.
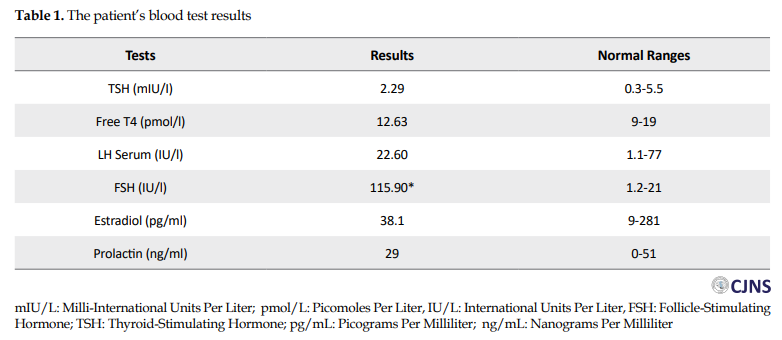
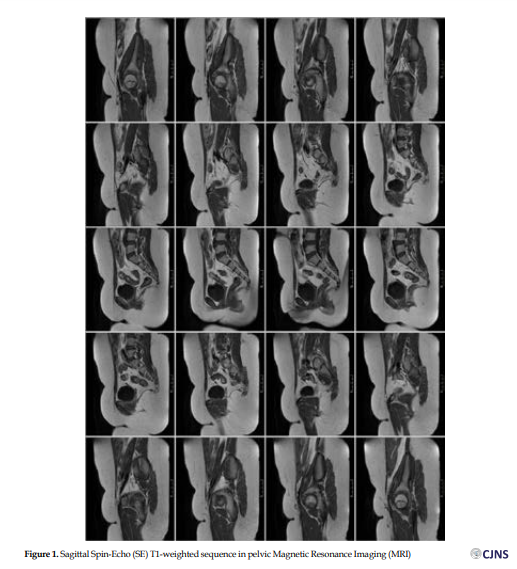
Her karyotype indicated normal female 46, XX. Follicle-Stimulating Hormone (FSH) level was above normal. All other hormone levels were within normal limitations (Table 1). In addition, on non-contrast abdominopelvic MRI, no uterus or cervix was noted. No ovarian structure could be found, as well. The vaginal length was normal. The bladder and muscular and bony structures appeared unremarkable. No ascites or pathologic lymphadenopathy were present (Figures 1 and 2).
.PNG)
Discussion
Gonadal dysgenesis is the most common cause of primary amenorrhea. Early defects in the formation of the primordial follicle or differentiation of the ovary may result in gonadal dysgenesis. Chromosomal abnormalities in patients with gonadal dysgenesis range from microdeletion in the X chromosome to aneuploidy [7].
MRKH syndrome arises from an interruption in embryonic development of Mullerian ducts in an otherwise normal phenotype and normal 46, XX female karyotype. Genital tract abnormalities range from upper vaginal atresia to complete Mullerian agenesis and may be accompanied by urinary tract and/or skeletal anomalies [2].
Meticulous research, done using Google scholar and Pubmed, revealed 23 case reports (31 patients) since 2003 concerned with gonadal dysgenesis and MRKH co-occurrence (Table 2) [1-3, 8-27].
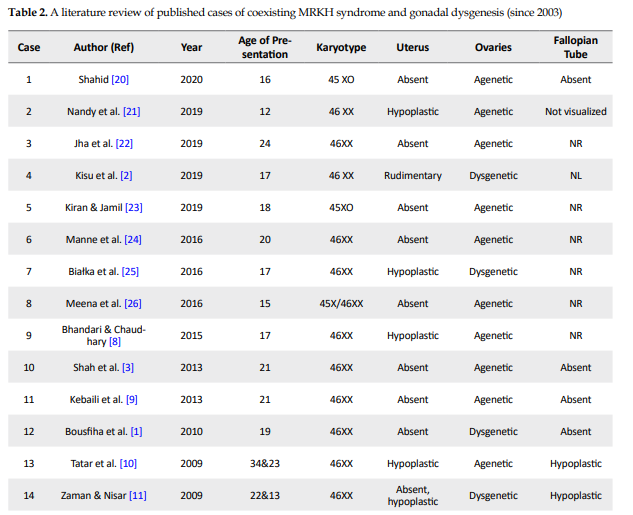
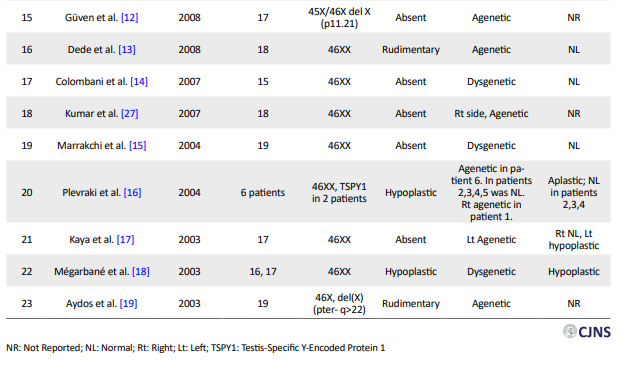
Almost all of them were presented in early adulthood with primary amenorrhea and underdeveloped secondary sexual features. Only two cases (cases 20 and 23) out of 31 were 45 XO and three (cases 12, 19, and 25) of them had specific deletions on chromosome X. Two karyotypes showed mosaicism (cases 2 and 26) and the rest were 46XX (cases 1–3, 8–11, 13–18, 21, 22, 24, and 27). Plevraki et al. studied six patients with MRKH syndrome and primary amenorrhea. Secondary sexual characteristics were well developed in all except patient number 6. Imaging revealed the presence of bilateral ovaries in patients number 2, 3, 4, and 5 and only the left ovary in patient number 1. The left fallopian tube was absent and the uterus was hypoplastic in patient number 1. The fallopian tubes were detected bilaterally but the uterus was hypoplastic in patients 2, 3, and 4.
The left fallopian tube was aplastic and the uterus was unicornuate in patient number 5. No gonadal tissue was identified in patient number 6 and the fallopian tubes and uterus were hypoplastic [16]. Considering the other 25 cases, the uterus was reported hypoplastic in six cases (cases 8, 10, 11, 18, 21, and 25), rudimentary in 3 cases (cases 2, 13, and 19) and absent in the rest (cases 1, 3, 24, 26, 27, 9, 12, 14, 15, 17, 20, 22, and 23). Ovaries were reported dysgenetic in seven cases (cases 1, 2, 11, 14, 15, 18, and 25) and agenetic in the rest (cases 3, 8, 22-24, 26, 27, 9, 10, 12, 13, 17, and 19-21). Although the state of fallopian tubes was not reported in some cases (cases 8, 12, 19, and 22–27), they were bilaterally normal in four cases (cases 2 and 13-15).
The current case differed from others to some extent. First, the coexisted brain anomaly was the Dandy-Walker variant, not the Dandy-Walker malformation, which was reported by Pillay et al. Second, the current patient underwent intestinal surgery at two days of age owing to duodenal atresia [28].
Duodenal atresia, the leading cause of inborn duodenal obstruction, seems to be the result of defective duodenal recanalization [29, 30]. It is frequently accompanied by conditions, such as Down’s syndrome, cardiac anomalies, VACTERL (vertebral, anorectal, tracheo-oesophageal, renal, and limb) association, annular pancreas, and malrotation [31, 32]. The incidence of duodenal atresia has been reported as 1 in 10,000 live births [33]. The simultaneous presence of duodenal atresia with gonadal dysgenesis and the Dandy-Walker variant has not been reported so far.
Conclusion
In this study, the authors observed that although the Mayer-Rokitansky-Küster-Hauser syndrome was limited to the genital system by definition, it may be associated with other congenital disorders, which necessitates the need for the assessment of other organs in these patients. Moreover, the pathogenesis of the coexistence of gonadal dysgenesis, MRKH syndrome, and the Dandy-Walker variant has not been identified so far. Although some affected chromosomal regions have been identified in this era, further genetic analyses should be performed to elucidate the probable association between these anomalies.
Ethical Considerations
Compliance with ethical guidelines
The study was approved by the ethics committee of Guilan University of Medical Sciences (Code: IR.GUMS.REC.1399.506). All ethical principles are considered in this article. The participants were informed about the purpose of the research and its implementation stages. They were also assured about the confidentiality of their information. They were free to leave the study whenever they wished, and if desired, the research results would be available to them.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Shahin Koohmanaee, Setila Dalili; Methodology: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Setila Dalili; Investigation: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Vahid Aminzadeh, Marjaneh Zarkesh, Seyyedeh Azade Hoseini Nouri, Fateme Rajaeipoor, Setila Dalili; Writing of the original draft: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Setila Dalili; Writing, review and editing: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Vahid Aminzadeh, Marjaneh Zarkesh, Seyyedeh Azade Hoseini Nouri, Fateme Rajaeipoor, Setila Dalili; Supervision: Shahin Koohmanaee, Setila Dalili.
Conflict of interest
The authors declared no conflicts of interests.
References
Gonadal dysgenesis is depicted by the absent or underdeveloped ovaries in females with primary amenorrhea and leads to variable degrees of hypogonadism and impuberism [1]. Patients with gonadal dysgenesis can have different karyotypes in the forms of 46 XX, 45 XO, mosaicism, or deletion of a specific part of the X chromosome [2].
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, the second most common cause of primary amenorrhea following gonadal dysgenesis, is related to the congenital absence or hypoplasia of the uterus and upper two-thirds of the vagina in a female with normal phenotype and karyotype. It affects nearly 1 out of 5,000 newborn girls [3]. MRKH syndrome can be whether isolated (type 1) or associated with renal and skeletal malformations, auditory defects, and occasionally, anomalies of the heart and digits (type 2) [4].
Dandy Walker Complex (DWC) is a Central Nervous System (CNS) malformation, which comprises of four types: 1- Dandy-Walker malformation marked by a triad of agenesis or hypoplasia of the cerebellar vermis, cystic dilatation of the fourth ventricle, and an enlarged posterior fossa; 2- Dandy-Walker variant associated with vermis hypoplasia, cystic dilatation of the fourth ventricle, and normal posterior fossa; 3- Mega cisternae Magna delineated by normal vermis and fourth ventricle but large posterior fossa; and 4- Posterior fossa arachnoid cyst [5].
Although the coexistence of gonadal dysgenesis and MRKH has been reported, it is still quite infrequent. To the extent that authors searched, just one study reported the association between Rokitansky sequence and Dandy-Walker malformation [6]. Here, the authors aimed to report a case with gonadal dysgenesis, MRKH, and the Dandy-Walker variant altogether.
Case Presentation
A 15-year-old girl was presented with primary amenorrhea. She had been experiencing two-day-lasting episodes of fatigue, lethargy, abdominal pain, and monthly vaginal discharge for two years.
Her birth weight was 2400 grams. She underwent intestinal surgery at two days of age because of excessive vomiting due to duodenal atresia. She was also hospitalized due to asthma and allergy four years before the recent attendance at this clinic. Moreover, due to blurred vision and repetitive drop attacks remaining for several months, brain Magnetic Resonance Imaging (MRI) was performed and revealed the Dandy-Walker variant. Subsequently, she was on piracetam, folic acid, and vitamin E for six months, which was followed by the resolution of symptoms and cessation of treatment four years before admission. The patient was the firstborn of first-degree cousin marriage. She was born preterm by Natural Vaginal Delivery (NVD). The mother had been pregnant five times, three of which led to NVD, and she had had two uninduced abortions.
On examination, the patient was in thelarche stage three and pubarche stage two. Abdominal obesity and striae were identified. Axillary hair was very sparse. She was 151 cm in height and 65 kg in weight with a Body Mass Index (BMI) of 28.5 (percentile= 95th). Her father’s and mother’s heights were 177 and 155 cm, respectively.
On pelvic transabdominal ultrasound, the uterus was not seen and both ovaries looked atrophic, small, and underdeveloped with no follicles, but the vagina was normal. The cervix and uterus could not be found. There was no evidence of free fluid in the pelvic cavity during both sonographies. Other organs seemed normal on ultrasound.
Her karyotype indicated normal female 46, XX. Follicle-Stimulating Hormone (FSH) level was above normal. All other hormone levels were within normal limitations (Table 1). In addition, on non-contrast abdominopelvic MRI, no uterus or cervix was noted. No ovarian structure could be found, as well. The vaginal length was normal. The bladder and muscular and bony structures appeared unremarkable. No ascites or pathologic lymphadenopathy were present (Figures 1 and 2).
Discussion
Gonadal dysgenesis is the most common cause of primary amenorrhea. Early defects in the formation of the primordial follicle or differentiation of the ovary may result in gonadal dysgenesis. Chromosomal abnormalities in patients with gonadal dysgenesis range from microdeletion in the X chromosome to aneuploidy [7].
MRKH syndrome arises from an interruption in embryonic development of Mullerian ducts in an otherwise normal phenotype and normal 46, XX female karyotype. Genital tract abnormalities range from upper vaginal atresia to complete Mullerian agenesis and may be accompanied by urinary tract and/or skeletal anomalies [2].
Meticulous research, done using Google scholar and Pubmed, revealed 23 case reports (31 patients) since 2003 concerned with gonadal dysgenesis and MRKH co-occurrence (Table 2) [1-3, 8-27].
Almost all of them were presented in early adulthood with primary amenorrhea and underdeveloped secondary sexual features. Only two cases (cases 20 and 23) out of 31 were 45 XO and three (cases 12, 19, and 25) of them had specific deletions on chromosome X. Two karyotypes showed mosaicism (cases 2 and 26) and the rest were 46XX (cases 1–3, 8–11, 13–18, 21, 22, 24, and 27). Plevraki et al. studied six patients with MRKH syndrome and primary amenorrhea. Secondary sexual characteristics were well developed in all except patient number 6. Imaging revealed the presence of bilateral ovaries in patients number 2, 3, 4, and 5 and only the left ovary in patient number 1. The left fallopian tube was absent and the uterus was hypoplastic in patient number 1. The fallopian tubes were detected bilaterally but the uterus was hypoplastic in patients 2, 3, and 4.
The left fallopian tube was aplastic and the uterus was unicornuate in patient number 5. No gonadal tissue was identified in patient number 6 and the fallopian tubes and uterus were hypoplastic [16]. Considering the other 25 cases, the uterus was reported hypoplastic in six cases (cases 8, 10, 11, 18, 21, and 25), rudimentary in 3 cases (cases 2, 13, and 19) and absent in the rest (cases 1, 3, 24, 26, 27, 9, 12, 14, 15, 17, 20, 22, and 23). Ovaries were reported dysgenetic in seven cases (cases 1, 2, 11, 14, 15, 18, and 25) and agenetic in the rest (cases 3, 8, 22-24, 26, 27, 9, 10, 12, 13, 17, and 19-21). Although the state of fallopian tubes was not reported in some cases (cases 8, 12, 19, and 22–27), they were bilaterally normal in four cases (cases 2 and 13-15).
The current case differed from others to some extent. First, the coexisted brain anomaly was the Dandy-Walker variant, not the Dandy-Walker malformation, which was reported by Pillay et al. Second, the current patient underwent intestinal surgery at two days of age owing to duodenal atresia [28].
Duodenal atresia, the leading cause of inborn duodenal obstruction, seems to be the result of defective duodenal recanalization [29, 30]. It is frequently accompanied by conditions, such as Down’s syndrome, cardiac anomalies, VACTERL (vertebral, anorectal, tracheo-oesophageal, renal, and limb) association, annular pancreas, and malrotation [31, 32]. The incidence of duodenal atresia has been reported as 1 in 10,000 live births [33]. The simultaneous presence of duodenal atresia with gonadal dysgenesis and the Dandy-Walker variant has not been reported so far.
Conclusion
In this study, the authors observed that although the Mayer-Rokitansky-Küster-Hauser syndrome was limited to the genital system by definition, it may be associated with other congenital disorders, which necessitates the need for the assessment of other organs in these patients. Moreover, the pathogenesis of the coexistence of gonadal dysgenesis, MRKH syndrome, and the Dandy-Walker variant has not been identified so far. Although some affected chromosomal regions have been identified in this era, further genetic analyses should be performed to elucidate the probable association between these anomalies.
Ethical Considerations
Compliance with ethical guidelines
The study was approved by the ethics committee of Guilan University of Medical Sciences (Code: IR.GUMS.REC.1399.506). All ethical principles are considered in this article. The participants were informed about the purpose of the research and its implementation stages. They were also assured about the confidentiality of their information. They were free to leave the study whenever they wished, and if desired, the research results would be available to them.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Shahin Koohmanaee, Setila Dalili; Methodology: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Setila Dalili; Investigation: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Vahid Aminzadeh, Marjaneh Zarkesh, Seyyedeh Azade Hoseini Nouri, Fateme Rajaeipoor, Setila Dalili; Writing of the original draft: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Setila Dalili; Writing, review and editing: Shahin Koohmanaee, Amirhossein Tamimi, Soroush Ahmadi Macciani, Atena Tamimi, Vahid Aminzadeh, Marjaneh Zarkesh, Seyyedeh Azade Hoseini Nouri, Fateme Rajaeipoor, Setila Dalili; Supervision: Shahin Koohmanaee, Setila Dalili.
Conflict of interest
The authors declared no conflicts of interests.
References
- Bousfiha N, Errarhay S, Saadi H, Ouldim K, Bouchikhi C, Banani A. Gonadal Dysgenesis 46, XX Associated with Mayer-Rokitansky-Kuster-Hauser Syndrome: One case report. Obstet Gynecol Int. 2010; 2010:847370. [DOI:10.1155/2010/847370] [PMID] [PMCID]
- Kisu I, Ono A, Iijma T, Katayama M, Iura A, Hirao N. Mayer-Rokitansky-Küster-Hauser syndrome with a uterine cervix and normal vagina associated with gonadal dysgenesis in a 46, XX female. J Obstet Gynaecol Res. 2019; 45(7):1386-90. [DOI:10.1111/jog.13956] [PMID]
- Shah VN, Ganatra PJ, Parikh R, Kamdar P, Baxi S, Shah N. Brief Communication Coexistence of gonadal dysgenesis and Mayer-Rokitansky-Kuster-Hauser syndrome in 46 , XX female : A case report and review of literature. Indian J Endocrinol Metab. 2013; 17(S 1):S274-7. [DOI:10.4103/2230-8210.119605] [PMID] [PMCID]
- Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-K € syndrome. Fertil Steril. 2011; 95(5):1589-94. [DOI:10.1016/j.fertnstert.2010.07.1062] [PMID]
- Jurcă MC, Kozma K, Petcheşi CD, Bembea M, Pop OL, Muţiu G, et al. Anatomic variants in dandy-walker complex. Rom J Morphol Embryol. 2017; 58(3):1051-55. [PMID]
- Illay KOP, Atthews LOSM, Ainwright HECW. Facio-auriculo-vertebral sequence in association with DiGeorge sequence, Rokitansky sequence, and Dandy-Walker malformation: Case report. Pediatr Dev Pathol. 2003; 6(4):355-60. [DOI:10.1007/s10024-003-1124-z] [PMID]
- Shah VN, Ganatra PJ, Parikh R, Kamdar P, Baxi S, Shah N. Coexistence of gonadal dysgenesis and Mayer-Rokitansky-Kuster-Hauser syndrome in 46, XX female: A case report and review of literature. Indian J Endocrinol Metab. 2013; 17(S 1):S274-7. [DOI:10.4103/2230-8210.119605] [PMID] [PMCID]
- Bhandari B, Chaudhary BK. Gonadal dysgenesis and the Mayer-RokitanskyKuster-Hauser syndrome in a girl with a 46, XX karyotype: A case report. Int J Contemp Pediatr. 2015; 2(3):246-8. [DOI:10.18203/2349-3291.ijcp20150537]
- Kebaili S, Chaabane K, Mnif MF, Kamoun M, Kacem FH, Guesmi N, et al. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser Syndrome in a girl with a 46, XX karyotype: A case report and review of literature. Indian J Endocrinol Metab. 2013; 17(3):505-8. [DOI:10.4103/2230-8210.111663] [PMID] [PMCID]
- Tatar A, Ocak Z, Tatar A, Yesilyurt A, Borekci B, Oztas S. Primary hypogonadism, partial alopecia, and Mullerian hypoplasia: Report of a third family and review. Am J Med Genet A. 2009; 149A(3):501-4. [DOI:10.1002/ajmg.a.32645] [PMID]
- Zaman SM, Nisar M. Primary hypergonadotrophic hypogonadism, alopecia totalis, and müllerian hypoplasia: A clinical study. J Pak Med Assoc. 2009; 59(8):571-3. [PMID]
- Güven A, Kara N, Sağlam Y, Güneş S, Okten G. The Mayer-Rokitansky-Kuster-Hauser and gonadal dysgenesis anomaly in a girl with 45, X/46,X,del(X)(p11.21). Am J Med Genet A. 2008; 146A(1):128-31. [DOI:10.1002/ajmg.a.32048] [PMID]
- Dede M, Gezginç K, Ulubay M, Alanbay I, Yenen M. A rare case of rudimentary uterus with absence of both ovaries and 46, XX normal karyotype without mosaicism. Taiwan J Obstet Gynecol. 2008; 47(1):84-6. [DOI:10.1016/S1028-4559(08)60060-1]
- Colombani M, Cau D, Thauvin-Robinet, Sapin E, Huet F, Faivre L. Fourth case of uterine aplasia, ovarian dysgenesis, amenorrhea and impuberism: A variant of Mayer-Rokitansky-Kuster-Hauser syndrome. Acta Paediatr. 2007; 96(9):1371-2. [DOI:10.1111/j.1651-2227.2007.00404.x] [PMID]
- Marrakchi A, Gharbi MH, Kadiri A. [Association dysgénésie gonadique et syndrome de Rokitansky Kuster Hauser: À propos d’un cas (French)]. Ann Endocrinol (Paris). 2004; 65(5):466-8. [DOI:10.1016/S0003-4266(04)95953-7]
- Plevraki E, Kita M, Goulis DG, Hatzisevastou-Loukidou H, Lambropoulos AF, Avramides A. Bilateral ovarian agenesis and the presence of the testis-specific protein 1-Y-linked gene: Two new features of Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril. 2004; 81(3):689-92. [DOI:10.1016/j.fertnstert.2003.07.029] [PMID]
- Kaya H, Sezik M, Özkaya O, Köse SA. Mayer-Rokitansky-Küster-Hauser syndrome associated with unilateral gonadal agenesis: A case report. J Reprod Med Obstet Gynecol. 2003; 48(11):902-4. [PMID]
- Mégarbané A, Gannagé-Yared MH, Khalifé AA, Fabre M. Primary hypergonadotropic hypogonadism, partial alopecia, and Müllerian hypoplasia: Report of a second family with additional findings. Am J Med Genet. 2003; 119A(2):214-7. [DOI:10.1002/ajmg.a.20170] [PMID]
- Aydos S, Tükün A, Bökesoy I. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser syndrome in a girl with 46, X, del(X) (pter→q22. Arch Gynecol Obstet. 2003; 267(3):173-4. [DOI:10.1007/s00404-001-0274-3] [PMID]
- Shahid MM. MRKH syndrome and Turner syndrome co-existing in a patient with primary amenorrhoea. Sri Lanka J Diabetes Endocrinol Metab. 2020; 10(1):30-3. [DOI:10.4038/sjdem.v10i1.7419]
- Nandy A, Naskar T, Datta D, Sardar P. Ovarian agenesis and Mullerian duct dysgenesis in a karyotypically normal (46, XX) pre-pubertal girl with aberrant cognition: A case report and literature review. Int J Med Rev Case Rep. 2019; 3(8):535-41. [DOI:10.5455/IJMRCR.Atypical-MRKH-syndrome-with-aberrant-cognition]
- Jha SK, Manandhar R, Shrivastava VR. Coexistence of gonadal dysgenesis and mullerian agenesis in a female with 46 XX karyotype: A case report. JNMA J Nepal Med Assoc. 2019; 57(216):119-22. [DOI:10.31729/jnma.4287]
- Kiran Z, Jamil T. Primary amenorrhoea secondary to two different syndromes: A case study. BMJ Case Rep. 2019; 12(3):e228148. [DOI:10.1136/bcr-2018-228148] [PMID] [PMCID]
- Manne S, Veeraabhinav CH, Jetti M, Himabindu Y, Donthu K, Badireddy M. A rare case of 46, XX gonadal dysgenesis and Mayer-Rokitansky-Kuster-Hauser syndrome. J Hum Reprod Sci. 2016; 9(4):263-6. [DOI:10.4103/0974-1208.197694] [PMID] [PMCID]
- Białka A, Gawlik A, Drosdzol-Cop A, Wilk K, Małecka-Tendera E, Skrzypulec-Plinta V. Coexistence of Mayer-Rokitansky-Küster-Hauser syndrome and Turner syndrome: A case report. J Pediatr Adolesc Gynecol. 2016; 29(2):e35-8. [DOI:10.1016/j.jpag.2015.10.019] [PMID]
- Meena A, Daga MK, Dixit R. Unusual association of turner syndrome and Mayer-Rokitansky-Küster-Hauser syndrome. BMJ Case Rep. 2016; 2016:bcr2015212634. [DOI:10.1136/bcr-2015-212634] [PMID] [PMCID]
- Kumar A, Mishra S, Dogra PN. Management of an unusual case of atypical Mayer-Rokitansky-Kuster-Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J Pelvic Floor Dysfunct. 2007; 18(7):823-5. [DOI:10.1007/s00192-006-0238-z] [PMID]
- Pillay K, Matthews LS, Wainwright HC. Facio-auriculo-vertebral sequence in association with DiGeorge sequence, Rokitansky sequence, and Dandy-Walker malformation: Case report. Pediatr Dev Pathol. 2003; 6(4):355-60. [DOI:10.1007/s10024-003-1124-z] [PMID]
- Mittal P, Peters NJ, Samujh R. A fatal combination of duodenal atresia with preduodenal portal vein, malrotation, and hypoplastic left heart syndrome: A case report. J Neonatal Surg. 2020; 9:19. [DOI:10.47338/jns.v9.550]
- O’rahilly R, Müller F. Human embryology & teratology. 2th ed. New York: Wiley-Liss; 1996. [DOI:10.1080/107710497175128]
- Choudhry MS, Rahman N, Boyd P, Lakhoo K. Duodenal atresia: Associated anomalies, prenatal diagnosis and outcome. Pediatr Surg Int. 2009; 25(8):727-30. [DOI:10.1007/s00383-009-2406-y] [PMID]
- Applebaum H, Sydorak R. Duodenal atresia and stenosis—annular pancreas. Coran AG, Caldamone A, Adzick NS, Krummel TM, Laberge JM, Shamberger R, editors. Pediatr Surgery (7th ed.). London: Elsevier Health Sciences; 2012. [DOI:10.1016/B978-0-323-07255-7.00081-7]
- Fonkalsrud EW, DeLorimier AA, Hays DM. Congenital atresia and stenosis of the duodenum. A review compiled from the members of the Surgical Section of the American Academy of Pediatrics. Pediatrics. 1969; 43(1):79-83. [PMID]
Type of Study: case report |
Subject:
Special
Received: 2021/07/7 | Accepted: 2021/08/29 | Published: 2021/10/1
Received: 2021/07/7 | Accepted: 2021/08/29 | Published: 2021/10/1
Send email to the article author
| Rights and permissions | |
 | This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
