


BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://cjns.gums.ac.ir/article-1-332-en.html



2- Pediatric Diseases Research Center, Guilan University of Medical Sciences, Rasht, Iran
Introduction
Inherited Metabolic Disorders (IMDs) are a class of genetic disorders. It commonly occurs as a result of single-gene defects which result in the defective function of gene products, especially enzymes. They are related to the toxic intermediary metabolites or decreased production of necessary metabolites [1].
Each metabolic disorder may have different types with various age of onset, clinical manifestations, severity, and even type of inheritance. IMDs manifest different signs and symptoms, including metabolic acidosis, nausea, and developmental delays. It can also have clinical presentations like cardiomegaly, skeletal dysplasia, ophthalmological disorders, and encephalopathies. However, the occurrence of these problems can be decreased through screening [2].
Although IMDs are generally detected in neonates or infants, they can be seen in adults too [3, 4]. Fortunately, newborn screening (NBS) tests can detect most of the congenital metabolic diseases [5]. Consanguinity with an estimated rate of 30% to 39% of marriages in Iran is one of the leading causes of IMD occurrence. In a consanguineous family, a higher rate of an autosomal recessive mutation can be transferred to the next generation [6]. Therefore, an increased risk of metabolic disorders and early mortality can be expected in these families [7].
So far, Iranian investigations have been conducted on the genetic disorders [8], especially IMDs, including biotinidase deficiency [9], methylmalonic academia [10], and non-ketotic hyperglycinemia [11]. These studies all showed the importance of considering metabolic disorders in our country. Ideally, a group of different specialists, including ophthalmologists, pediatricians, biochemists, and medical geneticists are needed for the final diagnosis and management of IMDs. So based on the abnormal metabolites direct toxic mechanisms on eyes [12] and regarding the effect of eye monitoring on follow-up, management, and treatment of IMDs [13], a detailed ophthalmological assessment is essential.
Because of the importance of the aforementioned issue, we aimed to demonstrate the effect of IMDs on eyes in this review study.
Materials and Methods
This narrative review article was conducted after a thorough electronic search by definite keywords. PubMed, Web of Science, Embase, and Google Scholar were searched to find the reference articles assessing the effect of IMDs on eyes.
Discussion
Eyes have anterior and posterior segments with different components. The cornea, sclera, iris, trabecular network, and lens are placed in the anterior segment. The posterior segment comprises the optic nerve, choroid, and retinal blood vessels [14]. Since the evaluation of an ophthalmologist along with biochemical and genetic evaluations could significantly help clinicians to diagnose and manage patients, it is wise to know ocular manifestations in IMDs. In this review article, the authors summarized the effect of ocular manifestations of IMDs by google images and only mentioned the effect of IMDs of amino acids, carbohydrates, and lipids on eyes.
Metabolic disorders can induce ocular abnormalities of conjunctiva, cornea, lens, retina, optic nerve, and ocular motility with symmetrical bilateral involvement and severe visual impairment.
Disorders of amino acid metabolism
Albinism, cystinosis, homocystinuria, tyrosinemia type II, hyperornithinemia (gyrate atrophy), sulfite oxidize deficiency, and hyperlysinemia are the most common disorders of amino acid metabolism. Albinism is an inherited disorder that causes hypopigmentation or no pigmentation in only eyes or a combination of skin, eyes, and hair [15]. Albinism causes refractive errors, nystagmus, decreased visual acuity, defects of iris transillumination, strabismus, albino fundus, and abnormal nerve fiber crossing [16].
Cystinosis is an inherited disorder that causes defective transport of cystine across lysosomal membranes. It creates corneal crystal accumulation, and pigmentary retinopathy in the eye [17].
Homocystinuria occurs because of the absence of cystathionine b-synthetase enzyme which converts homocysteine to cystathionine. Osteoporosis, fair skin, coarse hair, marfanoid habitus, mental retardation, convulsion, and thromboembolic episode are the systematic features of homocystinuria. Ocular manifestations of homocystinuria are retinal detachment, progressive myopia, ectopia lentis, increased intraocular pressure, and optic atrophy.
Sulfite oxidize deficiency occurs as a result of a defect in sulfur metabolism. It has some systemic features such as seizures, brain dysfunction (encephalopathy), spastic quadriplegia, movement problems, dystonia, choreoathetosis, ataxia, and ectopia lentis [18].
Hyperornithinemia (gyrate atrophy) is an autosomal recessive inherited chorioretinal dystrophy that occurs due to the deficiency of the ornithine aminotransferase which is the mitochondrial matrix enzyme activity. It has some ocular manifestations, including myopia, night blindness, chorioretinal atrophy, and cataract.
Tyrosinemia type II occurs because of mutations in the tyrosine aminotransferase. It is an autosomal recessive disorder and induces pseudodendritic keratitis, mental retardation, hyperkeratosis of the palms and soles. Hyperlysinemia is an extremely rare autosomal recessive disorder that causes mental retardation and ectopia lentis [12]. Ophthalmic manifestations of aminoacidopathies are shown in Figure 1.
.png)
Disorders of carbohydrate metabolism
Galactosemia, glycogen storage disease, glucose-6-phosphate dehydrogenase, and Von Gierke disease are the disorders of carbohydrate metabolism. But only galactosemia has ophthalmic manifestations. Galactosemia which is defined as the high level of galactose in the blood is a familial genetic disorder and is induced by the inability to metabolize galactose—the literal meaning of galactosemia. The different types of galactosemia include classic and clinical type, Duarte variant, galactokinase deficiency, epimerase deficiency [19]. Classic galactosemia which is defined by the defected galactose-l-phosphate uridyltransferase activity. Cataract may happen due to the accumulation of galactitol in the lens. It induces an oil drop appearance cataract (Figure 2).
.png)
Disorders of lipid metabolism
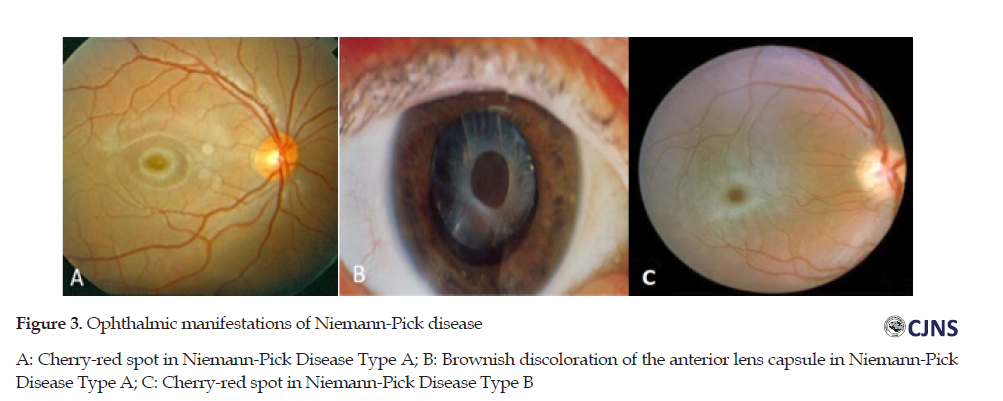
Niemann-pick, Fabry, Gaucher, metachromatic leukodystrophy (MLD), and gangliosidosis are the disorders of lipid metabolism. Niemann-Pick is an uncommon genetic disease. It disturbs the fat metabolism inside the cells such as cholesterol and lipids. Various organs, including eyes, can be affected by this disease. A, B, and C t are 3 types of Niemann-Pick disease [20]. There is various eye involvement in Niemann-Pick disease. The acute neuronopathic form is the Niemann-pick type A which produces a cherry-red spot in the macula. Another manifestation of this type of Niemann-Pick is the brownish discoloration of the anterior lens capsule. The chronic form of Niemann-Pick disease without nervous system involvement is type B which induces cherry-red spot in the macula. The chronic neuropathic form is type C. This type can have optic atrophy and vertical ophthalmoplegia. There is no cherry-red spot in this type [12]. Ophthalmic manifestations of Niemann-Pick disease are shown in Figure 3.
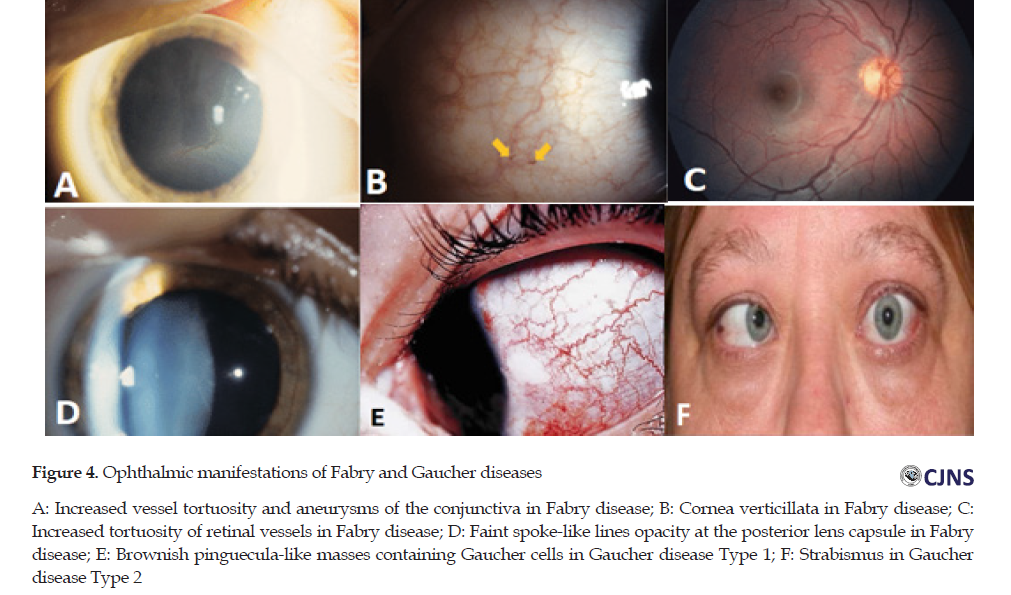
Fabry disease is a rare X-linked lysosomal storage disorder due to the mutations in the galactosidase alpha gene that causes the complete or partial deficiency of the enzyme α-galactosidase A, and mainly the subsequent slow accumulation of globotriaosylceramide [21]. Fabry disease causes the following ocular changes on conjunctiva, cornea, retina, and lens. The effects of the disease are increased vessel tortuosity and aneurysms on the conjunctiva, verticillata on the cornea, increased tortuosity of vessels on the retina, and faint spoke-like lines opacity at the posterior lens capsule on the lens. The most common ocular feature in all homozygotes and a majority (up to 70%) of heterozygotes is cornea verticillata that results in deposits of globotriaosylceramide (GB3) in corneal epithelium.
Gaucher disease is an uncommon, autosomal recessive disease that results from the deficient lysosomal enzyme (glucocerebrosidase). It leads to an accumulation of its substrate, glucosylceramide [22]. There are 3 types of Gaucher disease. The most common form is chronic, a non-neuropathic disorder with adult-onset (type 1). Its ocular features include brownish pinguecula-like masses containing Gaucher cells. In the retina, Gaucher cells can be observed as well. Gaucher disease type 2 is the acute neuropathic infantile form. It causes persistent retroflection of the head and signs of pseudobulbar palsy.
The classic Gaucher induces opisthotonus, trismus, and strabismus. The subacute, juvenile neuronopathic form is the Gaucher disease type 3 that has ocular features such as ocular apraxia and corneal opacification [23]. The ophthalmic manifestations of Fabry disease and Gaucher disease are shown in Figure 4.
MLD can be created by deficient activity of digesting sulfatides so their accumulation in MLD causes demyelination and inhibits oligodendrocyte differentiation from precursor cells, thereby preventing remyelination [24]. MLD is the accumulation of cerebroside sulfate in the CNS and peripheral nerves. Based on the age of onset, late infantile, juvenile, and adult forms are recognized. Multiple sulfatase deficiency (MSD) is an uncommon form of late-infantile MLD. Optic atrophy, retinal degeneration, skew deviation, and cherry-red spot induces ophthalmologic features [25].
The gangliosidosis diseases are IMDs in which the accumulation of ganglioside in the CNS leads to severe and progressive neurological impairment. Infantile gangliosidosis includes GM1 and GM2. GM2 gangliosidosis includes Tay-Sachs and Sandhoff diseases. Cherry red spot is the ocular manifestation of these disorders.
Conclusion
Because of the direct toxic mechanisms of abnormal metabolites on eyes and regarding the effect of eye monitoring on follow-up, management, and treatment of IMEs, a detailed ophthalmological assessment is essential.
Ethical Considerations
Compliance with ethical guidelines
All study procedures were in compliance with the ethical guidelines of the Declaration of Helsinki 2013.
Funding
This research received no specific grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
All authors contributed in preparing this article.
Conflict of interest
The authors declared no conflict of interest.
References
1.Yunus ZM, Rahman SA, Choy YS, Keng WT, Ngu LH. Pilot study of newborn screening of inborn error of metabolism using tandem mass spectrometry in Malaysia: Outcome and challenges. J Pediatr Endocrinol Metab. 2016; 29(9):1031-9. [DOI:10.1515/jpem-2016-0028] [PMID]
2.Sharma P, Gupta Sh, Kumar P, Sharma R, Mahapatra TK, Gupta G. Inborn error of metabolism screening in neonates. Natl J Physiol Pharm Pharmacol. 2019; 9(3):196-200. [DOI:10.5455/njppp.2019.9.1237608012019]
3.El-Hattab AW. Inborn errors of metabolism. Clin Perinatol. 2015; 42(2):413-39. [DOI:10.1016/j.clp.2015.02.010] [PMID]
4.Sedel F, Baumann N, Turpin JC, Lyon‐Caen O, Saudubray JM, Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis. 2007; 30(5):631-41. [DOI:10.1007/s10545-007-0661-4] [PMID]
5.Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH, et al. The tandem mass spectrometry newborn screening experience in North Carolina: 1997-2005. J Inherit Metab Dis. 2006; 29(1):76-85. [DOI:10.1007/s10545-006-0228-9] [PMID]
6.Erzurumluoglu AM, Shihab HA, Rodriguez S, Gaunt TR, Day INM. Importance of genetic studies in consanguineous populations for the characterization of novel human gene functions. Ann Hum Genet. 2016; 80(3):187-96. [DOI:10.1111/ahg.12150] [PMID] [PMCID]
7.Jouvet P, Touati G, Lesage F, Dupic L, Tucci M, Saudubray JM, et al. Impact of inborn errors of metabolism on admission and mortality in a pediatric intensive care unit. Eur J Pediatr. 2007; 166(5):461-5. [DOI:10.1007/s00431-006-0265-2] [PMID]
8.Dalili S, Bidabadi E, Behnam B. Bell’s palsy following growth hormone therapy in a patient with Prader-Willi syndrome: The first report. Genet Mol Res. 2018; 17(1):gmr16039877. https://www.geneticsmr.org/articles/bells-palsy-following-growth-hormone-therapy-in-a-patient-with-praderwilli-syndrome-the-first-report-7552.html
9.Koohmanaee Sh, Zarkesh M, Tabrizi M, Hassanzadeh Rad A, Divshali S, Dalili S. Biotinidase deficiency in newborns as respiratory distress and tachypnea: A case report. Iran J Child Neurol. 2015; 9(2):58-60. [DOI:10.22037/ijcn.v9i2.6137]
10.Karamizadeh Z, Dalili S, Karamifar H, Hossein Amirhakimi G. Association of methylmalonic acidemia and erythema nodosum. Iran J Med Sci. 2011;36(1):65-66.
11.Dalili S. Case presentation about non ketotic hyper glycinemia. Iran J Pediatr. 2014 Oct 1;24(S2):S11.
https://search.proquest.com/openview/a0380aff64c58bd0719f2b1c892c7ebd/1
12.Rajappa M, Goyal A, Kaur J. Inherited metabolic disorders involving the eye: A clinico-biochemical perspective. Eye. 2010; 24(4):507-18. [DOI:10.1038/eye.2009.229] [PMID]
13.Burlina A, Celato A, Burlina AP. Eye disorders. In: Hoffmann G, Zschocke J, Nyhan W, editors. Inherited Metabolic Diseases. Berlin/Heidelberg: Springer; 2017. p. 319-39. [DOI:10.1007/978-3-662-49410-3_30]
14.Irsch K, Guyton DL. Anatomy of eyes. In: Li SZ, Jain A, editors. Encyclopedia of Biometrics. Boston, MA: Springer; 2009. p. 1212-7. [DOI:10.1007/978-0-387-73003-5_253]
15.Fertl D, Rosel PE. Albinism. In: Perrin WF, Würsig B, Thewissen JGM, editors. Encyclopedia of Marine Mammals. Cambridge, MA: Academic Press; 2009. p. 24-6. [DOI:10.1016/B978-0-12-373553-9.00006-7]
16.Edralin SKR, Edwards M, Small DA, Williams R, Tornello I. Oculocutaneous albinism in embryonic development [Internet]. 2020 [Updated 2020 April 24]. Available from: https://scholarworks.boisestate.edu/under_showcase_2020/203/
17.Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: A review. Orphanet J Rare Dis. 2016; 11:47. [DOI:10.1186/s13023-016-0426-y] [PMID] [PMCID]
18.Sacharow SJ, Picker JD, Levy HL. Homocystinuria caused by cystathionine beta-synthase deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2017. https://www.ncbi.nlm.nih.gov/books/NBK1524/
19.Demirbas D, Coelho AI, Estela Rubio-Gozalbo M, Berry GT. Hereditary galactosemia. Metabolism. 2018; 83:188-96. [DOI:10.1016/j.metabol.2018.01.025] [PMID]
20.Eskes ECB, Sjouke B, Vaz FM, Goorden SMI, van Kuilenburg ABP, Aerts JMFG, et al. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol Genet Metab. 2020; 130(1):16-26. [DOI:10.1016/j.ymgme.2020.02.002] [PMID]
21.Svarstad E, Marti HP. The changing landscape of Fabry disease. Clin J Am Soc Nephrol. 2020; 15(4):569-76. [DOI:10.2215/CJN.09480819] [PMID]
22.Stirnemann J, Belmatoug N, Camou F, Serratrice Ch, Froissart R, Caillaud C, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017; 18(2):441. [DOI:10.3390/ijms18020441] [PMID] [PMCID]
23.Michalski A, Leonard JV, Taylor DSI. The eye and inherited metabolic disease: A review. J R Soc Med. 1988; 81(5):286-90.
[DOI:10.1177/014107688808100517] [PMID] [PMCID]
24.Wolf NI, Breur M, Plug B, Beerepoot Sh, Westerveld ASR, van Rappard DF, et al. Metachromatic leukodystrophy and transplantation: Remyelination, no cross‐correction. Ann Clin Transl Neurol. 2020; 7(2):169-80. [DOI:10.1002/acn3.50975] [PMID] [PMCID]
25.Jarnes Utz JR, Kim S, King K, Ziegler R, Schema L, Redtree ES, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab. 2017; 121(2):170-9. [DOI:10.1016/j.ymgme.2017.04.011] [PMID] [PMCID]
Received: 2020/09/6 | Accepted: 2020/04/22 | Published: 2020/07/1
| Rights and permissions | |
 | This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Owned by Guilan University of Medical Sciences.
Co-published by Negah Institute for Scientific Communication.
Contact Information
Caspian Journal of Neurological Sciences
Guilan University of Medical Sciences
University Tel : +9813 3333 0939
E-mail: cjnsgums@gmail.com, cjns@gums.ac.ir
