Wed, Apr 24, 2024
Volume 5, Issue 1 (Winter 2019)
Caspian J Neurol Sci 2019, 5(1): 41-47 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Esmaeili-Bandboni A, Davoudi A, Milani F, Afzali S, Sharafshah A, Fallahabadi F et al . A De Novo Deletion of Chromosome 18p with Persistent Limb Tremor and Difficulty Speaking. Caspian J Neurol Sci 2019; 5 (1) :41-47
URL: http://cjns.gums.ac.ir/article-1-251-en.html
URL: http://cjns.gums.ac.ir/article-1-251-en.html
Aghil Esmaeili-Bandboni1 

 , Arash Davoudi2 , Forozan Milani3 , Sara Afzali2 , Alireza Sharafshah2 , Fereshteh Fallahabadi2 , Parvaneh Keshavarz * 4
, Arash Davoudi2 , Forozan Milani3 , Sara Afzali2 , Alireza Sharafshah2 , Fereshteh Fallahabadi2 , Parvaneh Keshavarz * 4
, Arash Davoudi2 , Forozan Milani3 , Sara Afzali2 , Alireza Sharafshah2 , Fereshteh Fallahabadi2 , Parvaneh Keshavarz * 4
1- Department of Medical Genetics, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran
2- Division of Cytogenetic, Dr. Keshavarz Medical Genetics Lab, Rasht, Iran
3- Reproductive Health Research Center, Alzahra Hospital, Department of Obstetrics and Gynecology, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran
4- Division of Cytogenetic, Dr. Keshavarz Medical Genetics Lab, Rasht, Iran , keshavarz@gums.ac.ir
2- Division of Cytogenetic, Dr. Keshavarz Medical Genetics Lab, Rasht, Iran
3- Reproductive Health Research Center, Alzahra Hospital, Department of Obstetrics and Gynecology, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran
4- Division of Cytogenetic, Dr. Keshavarz Medical Genetics Lab, Rasht, Iran , keshavarz@gums.ac.ir
Full-Text [PDF 2991 kb]
(1339 Downloads)
| Abstract (HTML) (2444 Views)

Full-Text: (1082 Views)
Introduction
Macro-deletions of chromosome 18 (18p or 18q) occur in 1% of the live births [1]. Like other abnormalities in chromosome 18, the 18p deletion syndrome have a range of physical symptoms including variable mental retardation, growth retardation, low height, pectus excavatum, and craniofacial malformations such as long ear, ptosis, microcephaly and short neck. Until now, the number of reported cases with this syndrome were a little more than 150 cases [2-4]. At first, it was described in 1963 by a French researcher Jean de Grouchy [5]. So, it is also called as de Grouchy syndrome. Phenotypic variability of this syndrome makes it difficult to recognize [6].
In this case report study, we aimed to report an 18p deletion case with two new features. We tended to demonstrate the existence of this syndrome in our case using phenotypically features that had been observed in the previous studies and also using the cytogenetic examination of the patient.
Case Presentation
The patient is a 29-year-old girl. Written informed consent was obtained from the patient for the publication of this report. She is the third child of her parents with non-familial marriage, and without any history of similar cases in the family. The patient had been born full-term with a natural childbirth. The mother experienced no pregnancy problems, and had a normal delivery. After the birth, it was discovered that her baby was affected (blue and black face). Birth weight was within the normal range (3210 g). After birth and during her first years of life, she had tended to grow more slowly than her peers.
Up to nine months, she had reflux crisis. Then during childhood, it was necessary to keep her head high on the bed. Also, she snored noisily when asleep. The patient pronounced her first words at the age of three. There were the severe speech and language deficit in the patient. The patient was unable to state the obvious words. She was unable to go to the typical school, because of her low IQ (IQ score: 55-60). The puberty age of the patient was in 10.5 years old that is in the normal range [7-13].
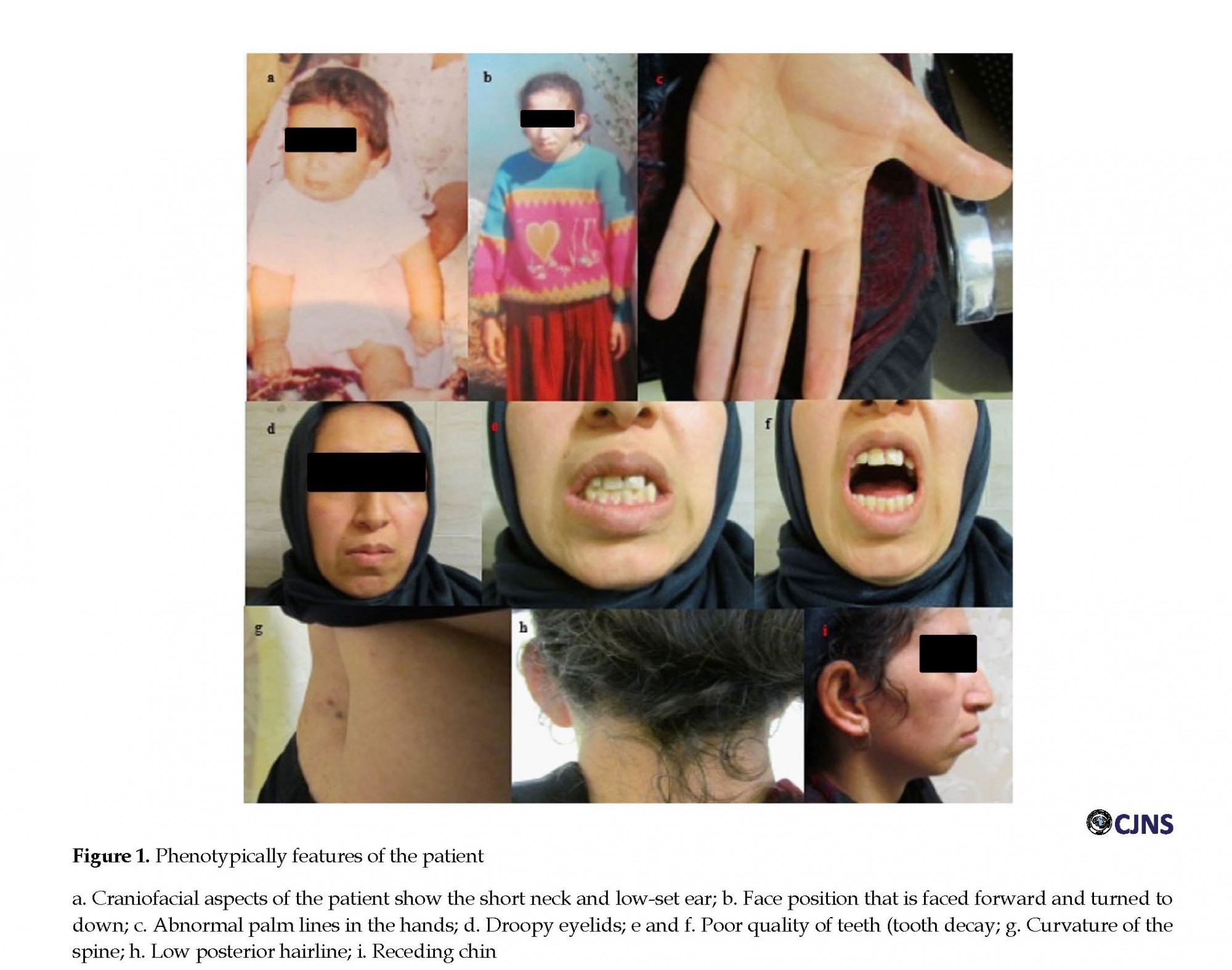
She also had difficulty in communicating with others. Her hand-eye coordination and motor skills were lower than her peers. The growth of her motor skills was delayed at least five months. During the physical exam at the age of 29, patient weight (52 kg) and height (155 cm) were somewhat lower than normal. She also has a short neck, that the head is faced forward (Figures 1 a and b). Furthermore, clinical studies on the patient showed that the palm lines on hands are not normal (Figure 1 c). Other facial features in the patient included; low- set ear (Figures 1 a, and b), low posterior hairline (Figure 1 h), curvature of the spine (kyphoscoliosis) (Figure 1 g), weak in ptosis (ability to open the eyelids fully) (Figure 1 d), and small and slightly receding chin and lower jaw (small mandible) (Figure 1 i). Furthermore, teeth investigation showed that the quality of the patient’s teeth is much lower than normal, with a large degree of decay and irregular dentition (Figures 1 e and f). In addition to, she is unable to pronounce the words correctly (difficulty speaking). Also, the patient has persistent tremor in her limbs that disturbs her lifestyle.
Analysis methods
Heparinized blood (5 mL) was used for this examination. Blood cells were cultured in RPMI 1640 (GIBCO, USA), supplemented by 20% (v/v) fetal bovine serum (GIBCO, USA) and 10 μg/ml phytohemagglutinin (GIBCO, USA) at 37°C. After 70-72 hours, 0.04 ng/mL of colchicine (Colchicine powder, Sigma Chemical Co., St. Louis, NJ, USA) was added to the culture. Peripheral blood lymphocytes were harvested by standard procedures [7]. The chromosomes were banded using the Giemsa-Trypsin-Giemsa (GTG) banding technique. Bands in twenty metaphase cells were evaluated under an Olympus CX31 microscope (New York Microscope Company, United States) with 450 bands/cell resolution [8].
Macro-deletions of chromosome 18 (18p or 18q) occur in 1% of the live births [1]. Like other abnormalities in chromosome 18, the 18p deletion syndrome have a range of physical symptoms including variable mental retardation, growth retardation, low height, pectus excavatum, and craniofacial malformations such as long ear, ptosis, microcephaly and short neck. Until now, the number of reported cases with this syndrome were a little more than 150 cases [2-4]. At first, it was described in 1963 by a French researcher Jean de Grouchy [5]. So, it is also called as de Grouchy syndrome. Phenotypic variability of this syndrome makes it difficult to recognize [6].
In this case report study, we aimed to report an 18p deletion case with two new features. We tended to demonstrate the existence of this syndrome in our case using phenotypically features that had been observed in the previous studies and also using the cytogenetic examination of the patient.
Case Presentation
The patient is a 29-year-old girl. Written informed consent was obtained from the patient for the publication of this report. She is the third child of her parents with non-familial marriage, and without any history of similar cases in the family. The patient had been born full-term with a natural childbirth. The mother experienced no pregnancy problems, and had a normal delivery. After the birth, it was discovered that her baby was affected (blue and black face). Birth weight was within the normal range (3210 g). After birth and during her first years of life, she had tended to grow more slowly than her peers.
Up to nine months, she had reflux crisis. Then during childhood, it was necessary to keep her head high on the bed. Also, she snored noisily when asleep. The patient pronounced her first words at the age of three. There were the severe speech and language deficit in the patient. The patient was unable to state the obvious words. She was unable to go to the typical school, because of her low IQ (IQ score: 55-60). The puberty age of the patient was in 10.5 years old that is in the normal range [7-13].
She also had difficulty in communicating with others. Her hand-eye coordination and motor skills were lower than her peers. The growth of her motor skills was delayed at least five months. During the physical exam at the age of 29, patient weight (52 kg) and height (155 cm) were somewhat lower than normal. She also has a short neck, that the head is faced forward (Figures 1 a and b). Furthermore, clinical studies on the patient showed that the palm lines on hands are not normal (Figure 1 c). Other facial features in the patient included; low- set ear (Figures 1 a, and b), low posterior hairline (Figure 1 h), curvature of the spine (kyphoscoliosis) (Figure 1 g), weak in ptosis (ability to open the eyelids fully) (Figure 1 d), and small and slightly receding chin and lower jaw (small mandible) (Figure 1 i). Furthermore, teeth investigation showed that the quality of the patient’s teeth is much lower than normal, with a large degree of decay and irregular dentition (Figures 1 e and f). In addition to, she is unable to pronounce the words correctly (difficulty speaking). Also, the patient has persistent tremor in her limbs that disturbs her lifestyle.
Analysis methods
Heparinized blood (5 mL) was used for this examination. Blood cells were cultured in RPMI 1640 (GIBCO, USA), supplemented by 20% (v/v) fetal bovine serum (GIBCO, USA) and 10 μg/ml phytohemagglutinin (GIBCO, USA) at 37°C. After 70-72 hours, 0.04 ng/mL of colchicine (Colchicine powder, Sigma Chemical Co., St. Louis, NJ, USA) was added to the culture. Peripheral blood lymphocytes were harvested by standard procedures [7]. The chromosomes were banded using the Giemsa-Trypsin-Giemsa (GTG) banding technique. Bands in twenty metaphase cells were evaluated under an Olympus CX31 microscope (New York Microscope Company, United States) with 450 bands/cell resolution [8].
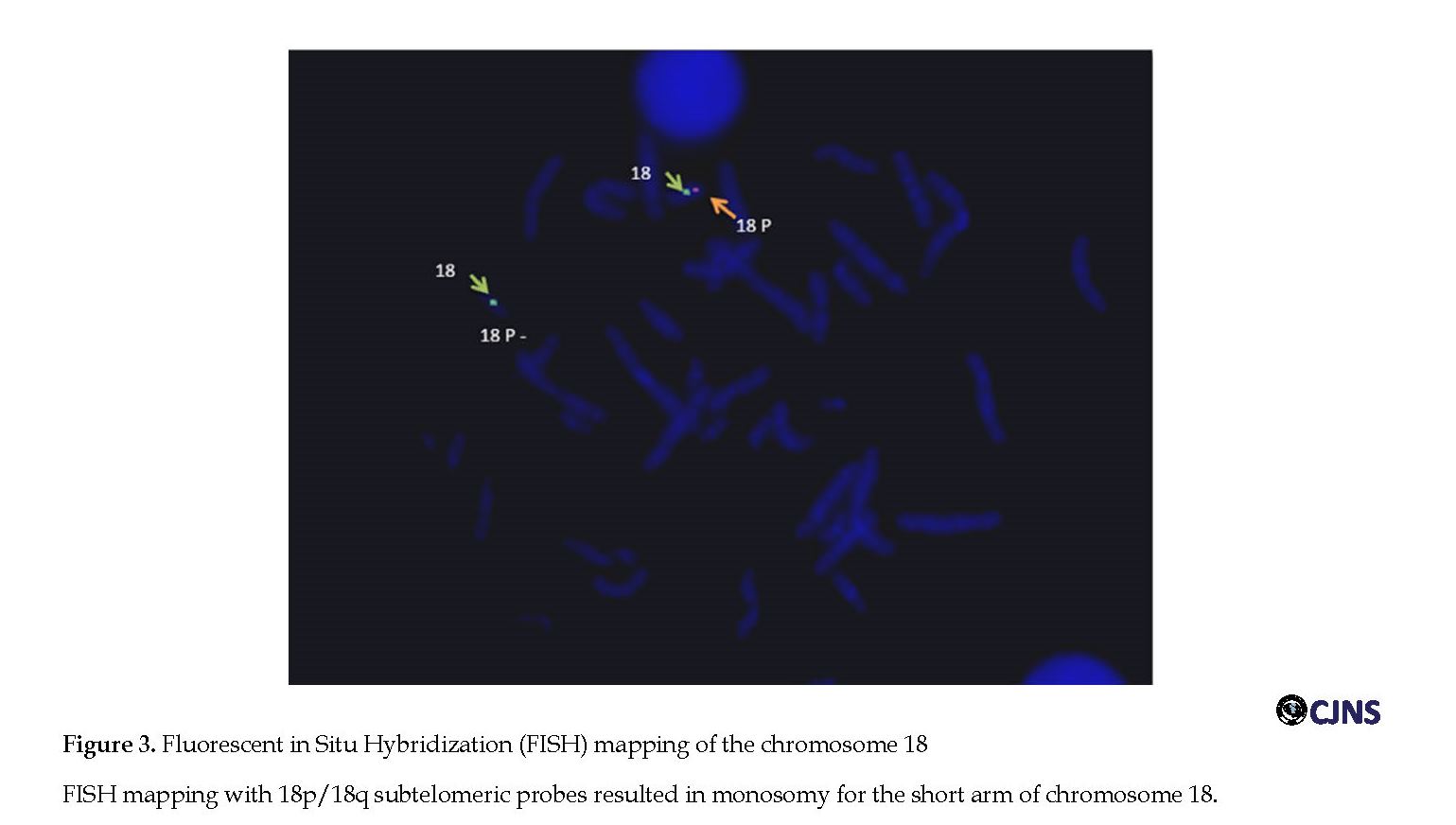
Chromosomal analysis (classified according to the ISCN [9]) revealed a deletion involving the short arm of chromosome 18: 46, XX, del(18)(p11.2) (Figure 2). The karyotypes of the parents were normal. Further investigation, Fluorescence in Situ Hybridization (FISH) assays with 18p Subtelomere (Cy5) FISH Probe (Taipei City, Taiwan, Catalog no: FE0160) and CEN18p (FITC) FISH Probe (Taipei City, Taiwan, Catalog no: FC0141) on 25 mitoses, was in agreement with cytogenetic results and showed a terminal deletion (about 9 Mbp) at 18p (Figure 3).
Discussion
Spontaneous (De Novo) errors with unknown reasons that are performed in the early stages of embryo development are the common causes of the 18p deletion syndrome [10]. Deletion of chromosome 18p has other appellations such as monosomy 18p, 18p deletion syndrome, and de Grouchy syndrome [11]. The study of clinical features of deletion p can help early and optimized diagnosis.
Spontaneous (De Novo) errors with unknown reasons that are performed in the early stages of embryo development are the common causes of the 18p deletion syndrome [10]. Deletion of chromosome 18p has other appellations such as monosomy 18p, 18p deletion syndrome, and de Grouchy syndrome [11]. The study of clinical features of deletion p can help early and optimized diagnosis.
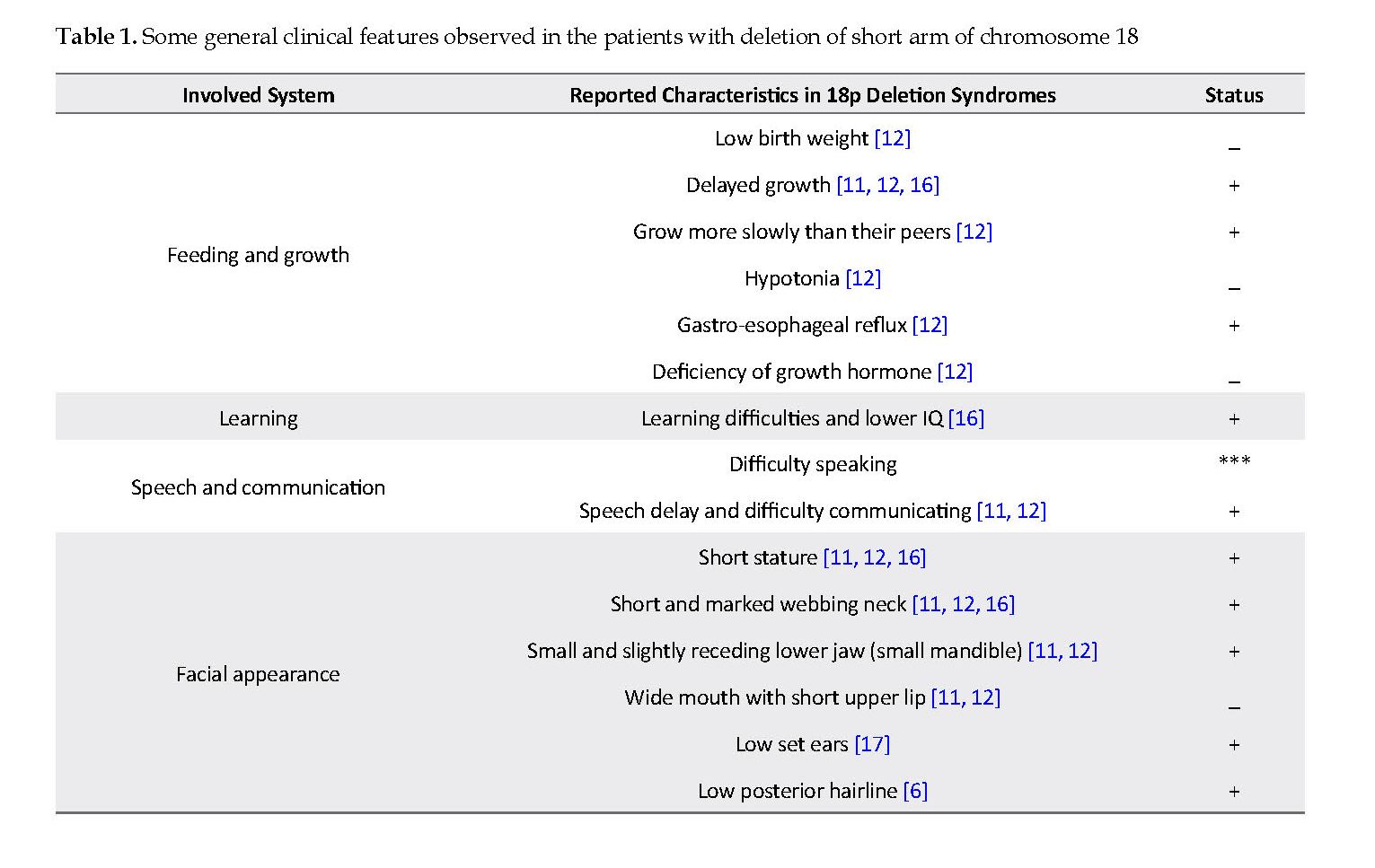
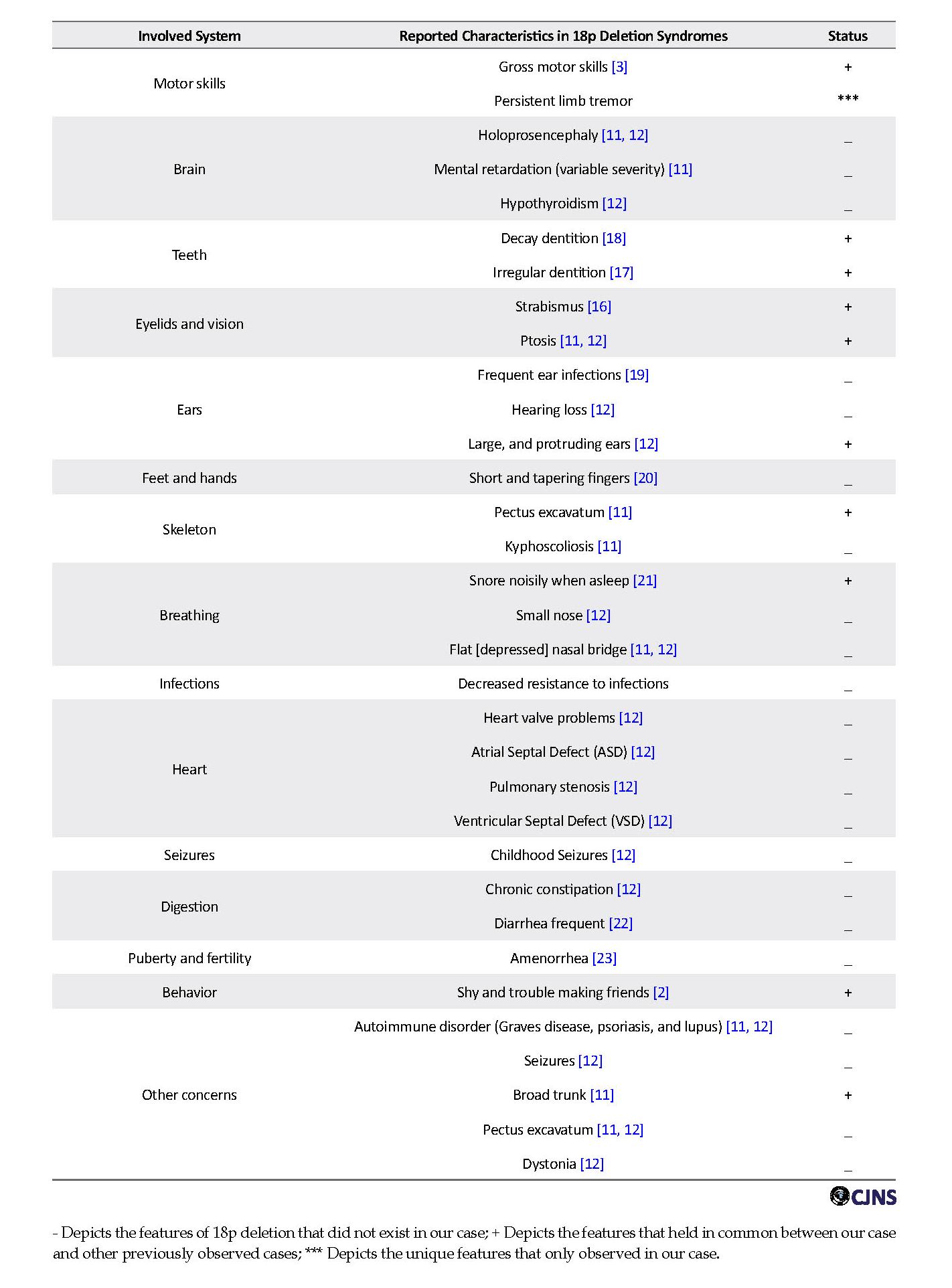
This paper presented a case of 18p deletion. Deleted part of 18p consists many genes such as PTPN2 (Tyrosine-protein phosphatase non-receptor type 2), PTPRM (Tyrosine-protein phosphatase, receptor type M), TGIF1 (TG-interacting factor 1) and SMCHD1 (Structural maintenance of chromosome flexible hinge domain containing 1). Many phenotypical features of this case were very similar to the other reported cases that listed out in Table 1 [3, 6, 12, 13]. Our case characteristically was presented with delayed and slowly growth, gastro-esophageal reflux, learning difficulties, postponement of speech, difficulty communicating, short stature, short neck, small and slightly receding chin or lower jaw, low-set ear, low posterior hairline, gross motor skills, decay and irregular dentition, ptosis, and scoliosis. Also in this case, two unique features were observed that were not seen in the previous studies [3, 12, 14, 15]; difficulty speaking, and persistent limb tremor.
This girl has a lot of problems to pronounce the words, correctly. This problem is so severe to the extent that she cannot state any obvious word. The other unique feature in our case is the persistent tremor in 4 limbs. Physical movements in this patient are always unbalanced. These recent two features are unique for our case than the previous cases of 18p deletion.
Conclusion
As a general conclusion, we found two new features for the diagnosis of patients with deletion of the short arm of chromosome 18. We hope that these two features can help in the optimized diagnosis of 18p syndromes. According to the knowledge that parents of our patient had normal karyotyping and might be performed abnormalities in gametogenesis of parents, the possibility of girl’s chromosome rearrangement involving 18p deletion is great. Therefore, we suggest parents who have a child with 18p deletion should note the possibility of having another pregnancy with an 18p deletion depends on the chromosomes of their gametes. Also, we suggest the novel molecular methods such as DNA microarray, and Real-Time PCR can be useful for precise detection of genes that localized in the short arm of chromosome 18.
Ethical Considerations
Compliance with ethical guidelines
Written informed consent was obtained from the patient for the publication of this report. All the study procedures were in compliance with the ethical guidelines of the Declaration of Helsinki 1957.
Funding
This study was supported by an intra-institution grant provided by the dean of Medical Genetic Laboratory of Dr. Keshavarz, Guilan, Iran.
Authors contributions
Experimental research and analysis: Sara Afzali, Alireza Sharafshah, Fereshteh Fallahabadi; Draft: Aghil Esmaeili-bandboni1, Arash Davoudi , Forozan Milani; Writing-review & editing: All authors; Funding acquisition: Parvaneh Keshavar; Resources: All authors; and Supervision: Parvaneh Keshavar, Forozan Milani.
Conflict of interest
The authors declared no conflict of interest.
References
Zavala J, Ramirez M, Medina R, Heard P, Carter E, Crandall A, et al. Psychiatric syndromes in individuals with chromosome 18 abnormalities. Am J Med Genet B Neuropsychiatr Genet. 2010; 153(3):837-45. [DOI:10.1002/ajmg.b.31047]
Ghaziuddin M, Sheldon S, Tsai L, Alessi N. Abnormalities of chromosome 18 in a girl with mental retardation and autistic disorder. J Intellect Disabil Res. 1993; 37(3):313-7. [DOI:10.1111/j.1365-2788.1993.tb01288.x]
Klein C, Page C, LeWitt P, Gordon M, De Leon D, Awaad Y, et al. Genetic analysis of three patients with an 18p− syndrome and dystonia. Neurol. 1999; 52(3):649. [DOI:10.1212/WNL.52.3.649]
Postma AG, Verschuuren-Bemelmans CC, Kok K, van Laar T. Characteristics of dystonia in the 18p deletion syndrome, including a new case. Clin Neurol Neurosurg. 2009; 111(10):880-2. [DOI:10.1016/j.clineuro.2009.07.013]
De Grouchy J. [Complex dysmorphism with oligophrenia. Deletion of the short arms of chromosome 17-18 (French)]. CR Acad Sci Paris. 1963; 256:1028-9.
Maranda B, Lemieux N, Lemyre E. Familial deletion 18p syndrome: Case report. BMC Med Genet. 2006; 7:60. [DOI:10.1186/1471-2350-7-60]
Lawce HJ, Brown MG. Peripheral blood cytogenetic methods. In: Arsham MS, Barch MJ, Lawce HJ, Brown MG, editors. The AGT cytogenetics laboratory manual. Hoboken, New Jersey: Wiley; 2017. [DOI:10.1002/9781119061199.ch3]
Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971; 298(7731):971-2. [DOI:10.1016/S0140-6736(71)90287-X]
Shaffer LG, McGowan-Jordan J, Schmid M. ISCN 2013: An international system for human cytogenetic nomenclature (2013). Basel: Karger Medical and Scientific Publishers; 2013.
Kasasbeh FA, Shawabkeh MM, Hawamdeh AA. Deletion of 18p syndrome. Lab Med. 2011; 42(7):436-8. [DOI:10.1309/LMAPLK2TVJBX5K9M]
Turleau C. Monosomy 18p. Orphanet J Rare Dis. 2008; 3:4. [DOI:10.1186/1750-1172-3-4]
Sebold C, Soileau B, Heard P, Carter E, O’donnell L, Hale DE, et al. Whole arm deletions of 18p: Medical and developmental effects. Am J Med Genet A. 2015; 167(2):313-23. [DOI:10.1002/ajmg.a.36880]
Chen CP, Lin SP, Chern SR, Wu PS, Chen SW, Lai ST, et al. A 13-year-old girl with 18p deletion syndrome presenting Turner syndrome-like clinical features of short stature, short webbed neck, low posterior hair line, puffy eyelids and increased carrying angle of the elbows. Taiwan J Obstet Gynecol. 2018; 57(4):583-7. [DOI:10.1016/j.tjog.2018.06.019]
Graziadio C, Rosa RFM, Zen PRG, Pinto LLdC, Barea LM, Paskulin GA. Dystonia, autoimmune disease and cerebral white matter abnormalities in a patient with 18p deletion. Arq Neuropsiquiatr. 2009; 67(3A):689-91. [DOI:10.1590/S0004-282X2009000400021]
Pachajoa H, Saldarriaga W, Isaza C. 18p-syndrome: Presentation of two cases with alobar holoprosencenphaly. Colomb Med. 2010; 41(4):367-72.
Van Dyke HE, Valdmanis A, Mann JD. Probable deletion of the short arm of chromosome 18. Am J Hum Genet. 1964; 16(3):364-74. [PMCID]
Wester U, Bondeson ML, Edeby C, Annerén G. Clinical and molecular characterization of individuals with 18p deletion: a genotype–phenotype correlation. Am J Med Genet A. 2006; 140(11):1164-71. [DOI:10.1002/ajmg.a.31260]
Hermesch CB, Cody JT, Cody JD. Dental caries history in nine children with chromosome 18p deletion syndrome. Spec Care Dentist. 2000; 20(2):53-5. [DOI:10.1111/j.1754-4505.2000.tb01143.x]
Nazarenko SA, Ostroverkhova NV, Vasiljeva EO, Nazarenko LP, Puzyrev VP, Malet P, et al. Keratosis pilaris and ulerythema ophryogenes associated with an 18p deletion caused by a Y/18 translocation. Am J Med Genet. 1999; 85(2):179-82. [DOI:10.1002/(SICI)1096-8628(19990716)85:23.0.CO;2-R]
Takeda K, Okamura T, Hasegawa T. Sibs with tetrasomy 18p born to a mother with trisomy 18p. J Med Genet. 1989; 26(3):195-7. [DOI:10.1136/jmg.26.3.195]
Abusrewil S, McDermott A, Savage D. Growth hormone, suspected gonadotrophin deficiency, and ring 18 chromosome. Arch Dis Child. 1988; 63(9):1090-1. [DOI:10.1136/adc.63.9.1090]
Czakó M, Riegel M, Morava É, Schinzel A, Kosztolányi G. Patient with rheumatoid arthritis and MCA/MR syndrome due to unbalanced der (18) transmission of a paternal translocation t (18; 20)(p11. 1; p11. 1). Am J Med Genet. 2002; 108(3):226-8. [DOI:10.1002/ajmg.10243]
Stoffer SS, Koen AL, Abbasi AA, Brown S, Opitz JM. 46, XX, del (18p) with amenorrhea, hypothyroidism, and ptosis. Am J Med Genet. 1981; 9(4):285-90. [DOI:10.1002/ajmg.1320090404]
As a general conclusion, we found two new features for the diagnosis of patients with deletion of the short arm of chromosome 18. We hope that these two features can help in the optimized diagnosis of 18p syndromes. According to the knowledge that parents of our patient had normal karyotyping and might be performed abnormalities in gametogenesis of parents, the possibility of girl’s chromosome rearrangement involving 18p deletion is great. Therefore, we suggest parents who have a child with 18p deletion should note the possibility of having another pregnancy with an 18p deletion depends on the chromosomes of their gametes. Also, we suggest the novel molecular methods such as DNA microarray, and Real-Time PCR can be useful for precise detection of genes that localized in the short arm of chromosome 18.
Ethical Considerations
Compliance with ethical guidelines
Written informed consent was obtained from the patient for the publication of this report. All the study procedures were in compliance with the ethical guidelines of the Declaration of Helsinki 1957.
Funding
This study was supported by an intra-institution grant provided by the dean of Medical Genetic Laboratory of Dr. Keshavarz, Guilan, Iran.
Authors contributions
Experimental research and analysis: Sara Afzali, Alireza Sharafshah, Fereshteh Fallahabadi; Draft: Aghil Esmaeili-bandboni1, Arash Davoudi , Forozan Milani; Writing-review & editing: All authors; Funding acquisition: Parvaneh Keshavar; Resources: All authors; and Supervision: Parvaneh Keshavar, Forozan Milani.
Conflict of interest
The authors declared no conflict of interest.
References
Zavala J, Ramirez M, Medina R, Heard P, Carter E, Crandall A, et al. Psychiatric syndromes in individuals with chromosome 18 abnormalities. Am J Med Genet B Neuropsychiatr Genet. 2010; 153(3):837-45. [DOI:10.1002/ajmg.b.31047]
Ghaziuddin M, Sheldon S, Tsai L, Alessi N. Abnormalities of chromosome 18 in a girl with mental retardation and autistic disorder. J Intellect Disabil Res. 1993; 37(3):313-7. [DOI:10.1111/j.1365-2788.1993.tb01288.x]
Klein C, Page C, LeWitt P, Gordon M, De Leon D, Awaad Y, et al. Genetic analysis of three patients with an 18p− syndrome and dystonia. Neurol. 1999; 52(3):649. [DOI:10.1212/WNL.52.3.649]
Postma AG, Verschuuren-Bemelmans CC, Kok K, van Laar T. Characteristics of dystonia in the 18p deletion syndrome, including a new case. Clin Neurol Neurosurg. 2009; 111(10):880-2. [DOI:10.1016/j.clineuro.2009.07.013]
De Grouchy J. [Complex dysmorphism with oligophrenia. Deletion of the short arms of chromosome 17-18 (French)]. CR Acad Sci Paris. 1963; 256:1028-9.
Maranda B, Lemieux N, Lemyre E. Familial deletion 18p syndrome: Case report. BMC Med Genet. 2006; 7:60. [DOI:10.1186/1471-2350-7-60]
Lawce HJ, Brown MG. Peripheral blood cytogenetic methods. In: Arsham MS, Barch MJ, Lawce HJ, Brown MG, editors. The AGT cytogenetics laboratory manual. Hoboken, New Jersey: Wiley; 2017. [DOI:10.1002/9781119061199.ch3]
Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971; 298(7731):971-2. [DOI:10.1016/S0140-6736(71)90287-X]
Shaffer LG, McGowan-Jordan J, Schmid M. ISCN 2013: An international system for human cytogenetic nomenclature (2013). Basel: Karger Medical and Scientific Publishers; 2013.
Kasasbeh FA, Shawabkeh MM, Hawamdeh AA. Deletion of 18p syndrome. Lab Med. 2011; 42(7):436-8. [DOI:10.1309/LMAPLK2TVJBX5K9M]
Turleau C. Monosomy 18p. Orphanet J Rare Dis. 2008; 3:4. [DOI:10.1186/1750-1172-3-4]
Sebold C, Soileau B, Heard P, Carter E, O’donnell L, Hale DE, et al. Whole arm deletions of 18p: Medical and developmental effects. Am J Med Genet A. 2015; 167(2):313-23. [DOI:10.1002/ajmg.a.36880]
Chen CP, Lin SP, Chern SR, Wu PS, Chen SW, Lai ST, et al. A 13-year-old girl with 18p deletion syndrome presenting Turner syndrome-like clinical features of short stature, short webbed neck, low posterior hair line, puffy eyelids and increased carrying angle of the elbows. Taiwan J Obstet Gynecol. 2018; 57(4):583-7. [DOI:10.1016/j.tjog.2018.06.019]
Graziadio C, Rosa RFM, Zen PRG, Pinto LLdC, Barea LM, Paskulin GA. Dystonia, autoimmune disease and cerebral white matter abnormalities in a patient with 18p deletion. Arq Neuropsiquiatr. 2009; 67(3A):689-91. [DOI:10.1590/S0004-282X2009000400021]
Pachajoa H, Saldarriaga W, Isaza C. 18p-syndrome: Presentation of two cases with alobar holoprosencenphaly. Colomb Med. 2010; 41(4):367-72.
Van Dyke HE, Valdmanis A, Mann JD. Probable deletion of the short arm of chromosome 18. Am J Hum Genet. 1964; 16(3):364-74. [PMCID]
Wester U, Bondeson ML, Edeby C, Annerén G. Clinical and molecular characterization of individuals with 18p deletion: a genotype–phenotype correlation. Am J Med Genet A. 2006; 140(11):1164-71. [DOI:10.1002/ajmg.a.31260]
Hermesch CB, Cody JT, Cody JD. Dental caries history in nine children with chromosome 18p deletion syndrome. Spec Care Dentist. 2000; 20(2):53-5. [DOI:10.1111/j.1754-4505.2000.tb01143.x]
Nazarenko SA, Ostroverkhova NV, Vasiljeva EO, Nazarenko LP, Puzyrev VP, Malet P, et al. Keratosis pilaris and ulerythema ophryogenes associated with an 18p deletion caused by a Y/18 translocation. Am J Med Genet. 1999; 85(2):179-82. [DOI:10.1002/(SICI)1096-8628(19990716)85:23.0.CO;2-R]
Takeda K, Okamura T, Hasegawa T. Sibs with tetrasomy 18p born to a mother with trisomy 18p. J Med Genet. 1989; 26(3):195-7. [DOI:10.1136/jmg.26.3.195]
Abusrewil S, McDermott A, Savage D. Growth hormone, suspected gonadotrophin deficiency, and ring 18 chromosome. Arch Dis Child. 1988; 63(9):1090-1. [DOI:10.1136/adc.63.9.1090]
Czakó M, Riegel M, Morava É, Schinzel A, Kosztolányi G. Patient with rheumatoid arthritis and MCA/MR syndrome due to unbalanced der (18) transmission of a paternal translocation t (18; 20)(p11. 1; p11. 1). Am J Med Genet. 2002; 108(3):226-8. [DOI:10.1002/ajmg.10243]
Stoffer SS, Koen AL, Abbasi AA, Brown S, Opitz JM. 46, XX, del (18p) with amenorrhea, hypothyroidism, and ptosis. Am J Med Genet. 1981; 9(4):285-90. [DOI:10.1002/ajmg.1320090404]
Type of Study: case report |
Subject:
General
Received: 2018/05/17 | Accepted: 2018/10/28 | Published: 2019/01/1
Received: 2018/05/17 | Accepted: 2018/10/28 | Published: 2019/01/1
Send email to the article author
| Rights and permissions | |
 | This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Articles Copyright © The Author(s).
Owned by Guilan University of Medical Sciences.
Co-published by Negah Institute for Scientific Communication.
Contact Information
Caspian Journal of Neurological Sciences
Guilan University of Medical Sciences
University Tel : +9813 3333 0939
E-mail: cjnsgums@gmail.com, cjns@gums.ac.ir
Owned by Guilan University of Medical Sciences.
Co-published by Negah Institute for Scientific Communication.
Contact Information
Caspian Journal of Neurological Sciences
Guilan University of Medical Sciences
University Tel : +9813 3333 0939
E-mail: cjnsgums@gmail.com, cjns@gums.ac.ir
