

BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://cjns.gums.ac.ir/article-1-154-en.html


ABSTRACT
The B cell is a vital contributor to humoral immunity. The B cell-specific antigen CD20 is expressed during B cell development, starting at the pre-B cell level and persists through B cell differentiation, but is lost during terminal differentiation to plasma cells.
Rituximab is a monoclonal antibody that destroys both normal and malignant B cells that have CD20 on their surfaces and is therefore used to treat diseases characterized by excessive B cells, overactive B cells, or dysfunctional B cells.
The connective tissue diseases and vasculitis mediated by B cell may cause various disorders of the peripheral nervous system especially axonal neuropathy. B cell–directed therapies may represent a promising new treatment for autoimmune axonal neuropathies.
Keywords: B cell; Therapy; Giant Axonal Neuropathy; Mixed Connective Tissue Disease
Introduction
B cell is a vital contributor to humoral immunity. B cells belong to the group of white blood cells known as lymphocytes and can be distinguished from other subtypes of lymphocytes by the presence of B cell receptors on their surface. B cell receptors are membrane-bound immunoglobulins and represent the key molecules involved in B cell activation. The evolving B cell lineage includes pro-B cells, pre-B cells, immature and transitional B cells, mature naive B cells, memory B cells, plasmablasts, and plasma cells. Plasmablasts are recently differentiated antibody-producing cells that are usually short- lived but can re-circulate and settle in tissues such as the mucosa or the bone marrow, where they can differentiate into fully mature antibody-producing plasma cells [1,2]. The B cell-specific antigen CD20 is expressed during B cell development, starting at the pre-B cell level (not found on stem cells or early pre-B cells), and persists through B cell differentiation, but is lost during terminal differentiation to plasma cells [1].
Rituximab is a monoclonal antibody that destroys both normal and malignant B cells that have CD20 on their surfaces and is therefore used to treat diseases characterized by excessive B cells, overactive B cells, or dysfunctional B cells [2]. Three molecular features of CD20 made it an attractive choice for immunotherapy: (A) it does not internalize upon monoclonal antibody (mAb) binding; (B) it is not shed from the cell surface; (C) the physical association between CD20 and CD40 on the B cell surface enhances cell death [2,3]. Rituximab targets CD20 and if anti-CD40 mAb is added in the laboratory (not yet clinically available) additional cell death is noted [3]. B cells are not simply passive recipients of signals necessary for their differentiation into antibody-producing plasma cells. B cells behave as efficient antigen-presenting cells. Subsequently, a number of studies have described other functions of B cells, including the ability to promote T cell accumulation and activation to mediate antibody-independent autoimmune damage, and to provide co-stimulatory molecules and cytokines (e.g. TNF-a, interleukins-4 and -10) to sustain T cell activation. All these functions suggest B cells play a broader role in the pathogenesis of autoimmune diseases. After treatment with rituximab, for example, the repopulated B cells resemble those seen during normal ontogenesis as well as during repopulation after bone marrow transplantation, and suggests immune system resetting [2,4,5] (see Figure 1). Finally, although T- cell activation is considered to be a key component in the pathogenesis of many autoimmune diseases, recent evidence indicates that such activation depends on the presence of B cells [6].
Much of the available information about the mechanisms of B cell depletion has come from rituximab trials, with some additional insights from results of smaller controlled and uncontrolled studies involving the use of rituximab and other B cell–directed agents in patients with systemic lupus erythematosus (SLE). In human beings, administration of two 1000-mg doses of intravenous rituximab 2 weeks apart results in peak serum rituximab concentrations of 400 to 500 ug/mL. In patients with rheumatoid arthritis (RA), the mean distribution time for rituximab is 2.4 days, and the half-life is approximately 20 days. After two 1000-mg doses, measurable serum concentrations of rituximab (1-10 ug/mL) can be detected even after 6 months [5]. There is a relationship between serum rituximab concentrations and the degree of B cell depletion in patients with SLE [5]. In this study, there was a significant reduction in disease activity scores in subjects with SLE who depleted B cells to <5 cells/m, whereas subjects who did not deplete B cells to <5 cells/mL did not experience significant reductions in disease activity. Several studies have examined the relationships between circulating B cell subpopulations and disease response and relapse. After rituximab therapy, peripheral blood B cells begin to return in 6 to 9 months [5]. B cell recovery after rituximab therapy is associated with increased numbers of B cells with a plasma-blast phenotype including CD20. The absolute numbers of circulating blood memory B cells typically remain low for at least 1 year after rituximab therapy but gradually recover over time. In the best clinical responses, there were long-term reductions in peripheral blood and memory B cell populations and correspondingly high levels of peripheral blood transitional B cell populations [5]. However, if rituximab fails to control a certain autoimmune disease condition despite its ability to deplete B cells, it may be that certain CD20-B cells produce autoantibodies or play other critical roles in pathogenesis. In this regard, which B cell population rituximab depletes needs to be verified [7].
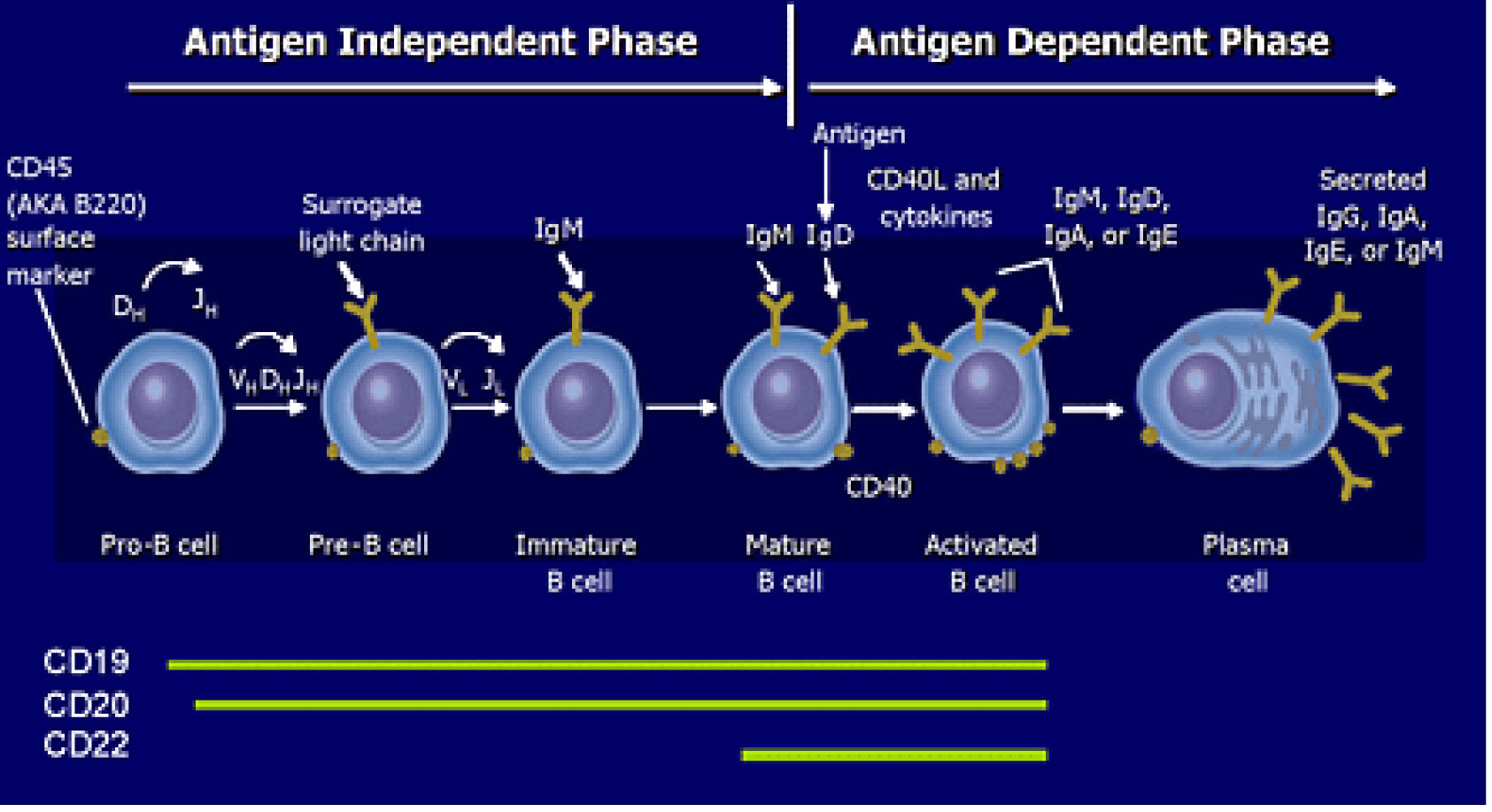
Figure 1. Targeting B Cell by CD surface marker in SLE. Liliane Min, M.D., Peter Barland, M.D., Elena Peeva, M.D. Department of Rheumatology, Albert Einstein College of Medicine, New York, NY 3 20 O6
A recent study indicates that CD20 plasma blasts may also play a critical role in a proportion of patients with RA who do not respond to anti‑CD20 antibodies. The study showed that amounts of IgJ mRNA, a marker for antibody‑secreting plasmablasts, are increased in such anti‑CD20 nonresponders. Which B cells should be targeted and how? This is a central question when retreating patients with B cell mediated autoimmune diseases [7]. Certain repopulated B cells may be more resistant to anti B cell therapy. These include B cells at a very early stage prior to antigen receptor selection (pre‑B cells).
Terminally differentiated antibody producing cells (long‑lived plasma cells) may not express CD20 on their surface [1]. As such, scheduled retreatment of RA with rituximab q 16 to 24 weeks would not wipe out all repopulated B cells [7] (see Figure 1).
Clinical studies of B cell–directed therapies are beginning to demonstrate the effects of specific interventions on immune function through delineation of B cell subsets by flow cytometry, measurement of serum autoantibodies and cytokines, and tests of immunocompetence. Most autoimmune disorders are characterized by the presence of autoantibodies and abnormalities of B-cell function [5]. Work has already identified B cell surface markers other than CD20 that may be targeted to improve the response. We know that treating B cells with both anti-CD20 and anti-CD40 antibodies together enhances the apoptotic response induced by a single dose of rituximab and is a more efficient therapeutic strategy for treating B cell-mediated disorders [3]. An antibody against another B cell marker, CD19, appears to remove a broader range of B cell populations, indicating that anti‑CD19 antibody therapy may be more effective in some diseases. However, the anti‑CD19 antibody does not deplete plasma cells in the bone marrow [7] (see Figure 1).
Mixed Connective Tissue Disease (MCTD)
MCTD is an example of a rare autoimmune disease, and gathering sufficient patients to study would be difficult. Patients manufacture their own unique autoantibodies. By definition, MCTD must have high titers of speckled-ANA and a positive anti-U1-RNP (formerly known as anti-ENA) antibody. Some individuals get a “fibromyalgia” syndrome while others develop a variable combination of neuropathy, polymyositis, congestive heart failure, interstitial pulmonary fibrosis, pulmonary arterial hypertension, or diffuse inflammatory (not erosive) arthritis. Double blinded placebo controlled studies of lupus, RA, and Sjögren’s syndrome have shown improved illness states unique to each autoimmune disease [8]. However, there is no evidence that elimination of CD20 B cells in MCTD treats the myopathy, neuropathy, arthritis, cardiomyopathy, or extensive fatigue that can complicate MCTD.
Although rituximab is not approved for the treatment of MCTD, CD4+ and CD8+ T cells from patients with active MCTD produce significantly more cytokines than cells in patients with inactive disease or in healthy individuals. Normal CD20 B cells produce these chemicals to help an immune system under attack by foreign antigens. However, the abnormal CD20 B cells of MCTD attack the person’s body even when there is no infection. This is the autoimmune response. MCTD arthritis evolves similarly to rheumatoid arthritis (RA) - for which Rituxan is approved - by attacking the CD20 B cells responsible for initiating the cascade of joint inflammation and destruction. Recent advances have led to the development of mAbs that effectively deplete B cells in human beings and target pathways essential for B cell development. Rituximab therapy produces variable decreases in both rheumatoid factor (RF) and anti-CCP (cyclic citrullinated peptide antibody) levels, irrespective of improvements in clinical disease activity [5]. The case appears to be similar for SLE, in which the autoantibodies react with double- stranded DNA and multiple ribonuclear proteins. Observed reductions in disease activity are not consistently correlated with decreases in these serum autoantibody levels [5]. This finding implies that B cells play an important role in RA and SLE beyond the production of autoantibodies, and that rituximab- induced B cell depletion acts through additional mechanisms as well. Among these agents, rituximab (Rituxan; Genentech, South San Francisco, California) has been the most studied in human beings.
Rituximab binds to CD20, which is almost exclusively expressed on human B cells [3,4]. Other mAbs that target B cells are in phase III of clinical development.
The patient stimulating this paper has MCTD with hand edema, classic nail-fold capillary changes, non-erosive arthritis, esophageal dysmotility, mild CHF and a progressive axonal neuropathy spanning several years but now presenting with progressive motor decline and gait ataxia from proprioceptive loss – with no other cerebellar symptoms or findings. There is no known treatment for MCTD, or the subset of diseases unique to each patient. He responded initially to methotrexate and high dose prednisone, but quickly developed methotrexate toxicity. He did not respond to a 6 month course of azathioprine and low dose prednisone (10mg daily). Rituximab was considered and the literature (i.e., PubMed) reviewed.
Axonal Neuropathy
This discussion is about axonal peripheral neuropathies, although there are other types of peripheral neuropathies that cause weakness. There are small and large fiber neuropathies. Unmyelinated C and thinly myelinated A∂ fibers are considered small fibers. Myelinated A∂ and Aß fibers are considered large fibers. Large fiber neuropathies can be axonal with affect proportional to nerve length (length dependent polyneuropathy). Demyelinating large fiber neuropathy affects the myelin sheath around axons and decreases the conduction speed of electrical impulses. This results in slow or no conduction (conduction block). Peripheral neuropathies can be both symmetrical and asymmetrical [9]
By nerve conduction criteria, 25% to 50% of SLE patients have neuropathy. In most, the neuropathy is a distal, symmetric, axonal type with mild clinical manifestations [8]. Although there are no descriptions of MCTD axonal neuropathy, chronic idiopathic axonal polyneuropathy (CIAP) has much in common with this patient’s neuropathy. CIAP refers to symmetrical, length dependant peripheral neuropathies where neurophysiology reveals axonal damage involving large fibers. The onset is insidious and shows slow or no progression over at least 6 months [9].
CIAP probably constitutes a heterogeneous group of conditions. Some studies suggest a role for the metabolic syndrome, which includes impaired glucose tolerance, dyslipidemia, hypertension and obesity. Our patient has none of the above. His MCTD suggests that autoimmunity plays a role. Autoimmunity is also suggested when CIAP pathological studies demonstrate an autoimmune vasculopathy [9]. Moreover, in other studies, some CIAP patients respond to immunosuppressive treatment and plasma exchange [9]. The concept of a primary autoimmune neuropathy, i.e., peripheral nerves being the primary target of an autoimmune response, has not been investigated but merits further consideration. It is always important to obtain a detailed medical history and perform a thorough physical examination to assess for underlying systemic autoimmune diseases that may be associated with an axonal neuropathy. [8]. The connective tissue diseases (SLE, RA, Sjögren’s syndrome, systemic sclerosis) and vasculitis may cause various disorders of the peripheral nervous system. The autoimmune disease with which MCTD is still most frequently confused is systemic lupus erythematosus (SLE). In fact, it is likely that several disorders can cause an axonal neuropathy [10]. The following outline will help identify the known causes of an axonal neuropathy:
- Symptoms can be divided into sensory and motor. Sensory symptoms included tingling, pins and needles, numbness, tightness, burning, pain and sensory ataxia. Motor symptoms included muscle cramps, stiffness, weakness andwasting.
- A history of comorbidities (i.e. diabetes), alcohol misuse, family history of neuropathy
- Check current medications - past and present – and a history of exposure to toxins
- Clinical examination and basic laboratory studies including:
- Hematology: CBC, Erythrocyte Sedimentation Rate, Vitamin B12, Folate
- Biochemistry: FBS, HbA1c, renal and liver functions, thyroid functions
- A coeliac disease screen
E. Finally, confirm the diagnosis and type of neuropathy with nerve conduction studies. Depending upon the findings and the patient’s desire, a referral to neurology would be appropriate. Other laboratory tests to be considered include: serum protein electrophoresis, serum angiotensin converting enzyme, oral glucose tolerance test, urinary Bence-Jones protein, ANA, RA, antineuronal antibodies, anti ENA, anti dsDNA, rheumatoid factor, p-ANCA, c-ANCA, Anti-HIV, and Anti-HCV. Imaging may be helpful: chest X-Ray, skeletal survey, abdominal and chest CT mammography, and PET scanning. Nerve biopsy may be needed after paraneoplastic screening; a thermal threshold test and a skin biopsy would complete the evaluation [9].
Axonal injury produces a typical pattern of abnormality on nerve conduction studies regardless of the cause. In most instances, axonal neuropathy is a chronic process. Changes caused by Wallerian degeneration may appear on nerve conduction study as early as 3 to 5 days after the onset of an acute axonopathy. Pure sensory axonopathies affect only sensory nerve amplitudes. Pure motor axonopathies affect only motor responses [11]. In the classic distal, symmetric, sensorimotor axonal neuropathy, there is initial loss of sensory nerve amplitude in a length-dependent fashion (i.e., first in the distal lower extremities) followed by loss of motor amplitudes, with gradual spread of these abnormalities to the shorter nerve segments in the upper extremities. This is largely because the more distal nerve segments in the legs are farther from their cell bodies (the anterior horn cells and dorsal root ganglia, in and near the spinal cord), which makes maintenance of the axon more difficult, increases its vulnerability to injury, and reduces its capacity to recover. Myelination is relatively preserved in primary axonal injury: distal latencies, conduction velocities, and late responses are not affected. Late in the course of severe axonal disorders (usually when amplitude has markedly decreased), these parameters may become mildly abnormal from secondary demyelination or loss of the fastest conducting fibers [11].
An EMG provides additional information when motor involvement is suspected in a patient with neuropathy. In a distal symmetric neuropathy, changes appear first distally and then move proximally as the neuropathy worsens and the deficits ascend. In severe acute processes, decreased motor unit recruitment and loss of a full interference pattern on voluntary contraction are the earliest indication of axon loss. With ongoing severe denervation, increased insertional activity and spontaneous activity appear, starting approximately 3 weeks after an acute injury, and may persist as long as the disease process remains active [11]. On EMG, motor unit activation (the motor unit action potential [MUAP]) generates larger MUAP wave-forms that have longer duration, higher amplitude, and increased complexity (neurogenic MUAPs). These neurogenic MUAPs are markers of chronic motor axon injury that do not appear for at least 2 to 3 months because of the time required for collateral reinnervation to become established [11]. Using these EMG abnormalities, it is possible to estimate the time since the onset of axonal injury and its severity. The degree of axonal loss is particularly important in patients with treatable neuropathies, because an estimate of axonal integrity allows an estimate of the potential for recovery of strength. Preservation of at least a moderate degree of axonal continuity (as indicated by a mild to moderate, rather than severe, reduction in the interference pattern on full voluntary contraction during EMG) suggests a good chance for further functional recovery, because these surviving axons provide the foundation for collateral reinnervation [11]. (NOTE: Guillain Barre’ syndrome is presented below as an example of an axonal neuropathy, but is not discussed above).
Autoantibody depletion correlates with the clinical effectiveness of these drugs in some but not all diseases. This suggests that much work needs to be done to understand the mechanism of action of these drugs. Recent studies correlating the clinical effectiveness of rituximab in patients with RA and SLE with depletion of B cells needs to be confirmed in larger cohorts of patients and needs to be studied in other autoimmune disorders [5].
Conclusions and Future Directions
B cell–directed therapies may represent a promising new treatment for autoimmune axonal neuropathies, although many questions remain about their optimal use. Any benefit of early treatment in an axonal neuropathy would not be known for months, as neurons die and regress slowly. Many patients would need to be recruited for a proper double blinded placebo controlled study to prove any benefit of early treatment. For example, one does not expect our patient to improve motor function once his neurons have died. Progressive motor decline after one treatment with rituximab will leave the patient at a lower but hopefully functional level that could be maintained with repeat treatments of rituximab. If any underlying contributing disease can be controlled, collateral reinnervation is often effective, bringing denervated muscle fibers back into service and restoring strength [11]. When denervating injury occurs slowly enough for collateral reinnervation to become established, nearly normal strength may be maintained even when up to half of the motor axons in a nerve have been lost [11]. However, impairments of muscle strength, sensory function and pain, fatigue and walking disability markedly interfere with patients’ functioning in daily life and adversely affect patient autonomy [12].
Conflict of Interest
The authors have no conflict of interest.
References
- Pateinakis P, Pyrpasopoulou A. CD20+ B Cell Depletion in Systemic Autoimmune Diseases: Common Mechanism of Inhibition or Disease-Specific Effect on Humoral Immunity? Biomed Res Int 2014; Article ID 973609. doi: 10.1155/2014/973609
- Perosa F, Prete M, Racanelli V, Dammacco F. CD20-depleting Therapy in Autoimmune Diseases: from Basic Research to the Clinic. J Intern Med 2010; 267(3):260-77. doi: 10.1111/j.1365-2796.2009.02207.x.
- Al-Zoobi L, Salti S, Colavecchio A, Jundi M, Nadiri A, Hassan GS, et al. Enhancement of Rituximab-induced Cell Death by the Physical Association of CD20 with CD40 Molecules on the Cell Surface. Int Immunol 2014; 26(8):451-65. doi: 10.1093/intimm/dxu046.
- Tak PP, Kalden JR. Advances in Rheumatology: New Targeted Therapeutics. Arthritis Res Ther 2011; 13(Suppl 1): S5. doi: 10.1186/1478-6354-13-S1-S5
- Levesque MC, St Clair EW. B Cell Directed Therapies for Autoimmune Disease and Correlates of Disease Response and Relapse. J Allergy Clin Immunol 2008; 121(1):13-21. doi: 10.1016/j.jaci.2007.11.030.
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B Cell Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis. N Engl J Med 2004; 350(25):2572-81.
- Yamamura T, Miyake S. B Cell Directed Therapy: Which B Cells Should be Targeted and How? Immunotherapy 2012; 4(5):455-7. doi: 10.2217/imt.12.35.
- Cojocaru IM, Cojocaru M, Silosi I, Vrabie CD. Peripheral Nervous System Manifestations in Systemic Autoimmune Diseases. Maedica (Buchar) 2014; 9(3):289-94.
- Zis P, Sarrigiannis PG, Rao DG, Hewamadduma C, Hadjivassiliou M. Chronic Idiopathic Axonal Polyneuropathy: A Systematic Review. J Neurol 2016; 263(10):1903-10. doi: 10.1007/s00415-016-8082-7.
- Hanewinckel R, van Oijen M, Ikram MA, van Doorn PA. The Epidemiology and Risk Factors of Chronic Polyneuropathy. Eur J Epidemiol 2016; 31(1):5-20. doi: 10.1007/s10654-015-0094-6.
- Gooch CL, Weimer LH. The Electrodiagnosis of Neuropathy: Basic Principles and Common Pitfalls. Neurol Clin 2007; 25(1):1-28.
- Erdmann PG, Teunissen LL, van Genderen FR, Notermans NC, Lindeman E, Helders PJ, et al. Functioning of Patients with Chronic Idiopathic Axonal Polyneuropathy (CIAP). J Neurol 2007; 254(9):1204-11.
Received: 2017/04/13 | Accepted: 2017/04/13 | Published: 2017/04/13
| Rights and permissions | |
 | This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Owned by Guilan University of Medical Sciences.
Co-published by Negah Institute for Scientific Communication.
Contact Information
Caspian Journal of Neurological Sciences
Guilan University of Medical Sciences
University Tel : +9813 3333 0939
E-mail: cjnsgums@gmail.com, cjns@gums.ac.ir
